-

-
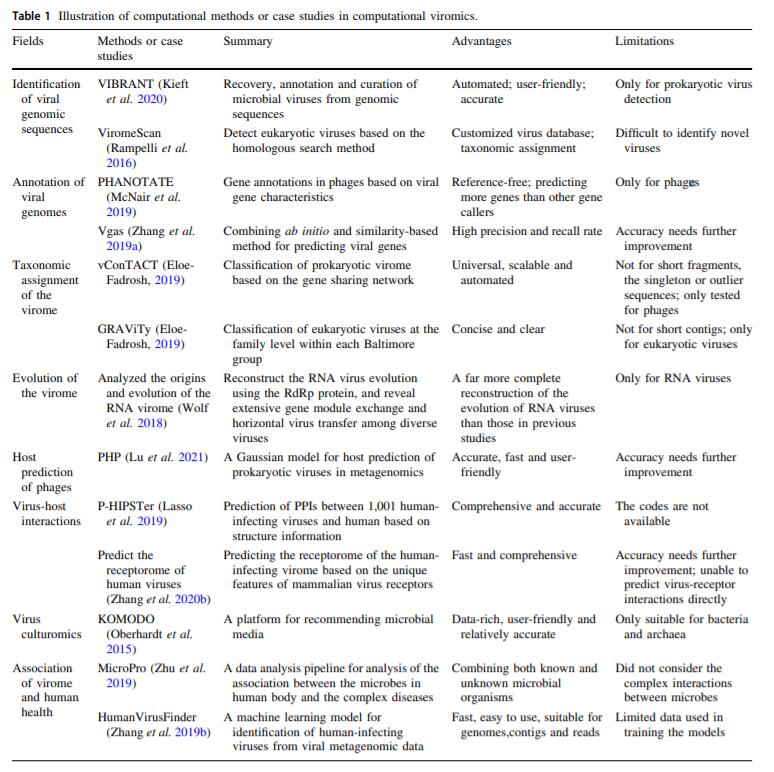
Fields Methods or case studies Summary Advantages Limitations Identification of viral genomic sequences VIBRANT (Kieft et al. 2020) Recovery, annotation and curation of microbial viruses from genomic sequences Automated; user-friendly; accurate Only for prokaryotic virus detection ViromeScan (Rampelli et al. 2016) Detect eukaryotic viruses based on the homologous search method Customized virus database; taxonomic assignment Difficult to identify novel viruses Annotation of viral genomes PHANOTATE (McNair et al. 2019) Gene annotations in phages based on viral gene characteristics Reference-free; predicting more genes than other gene callers Only for phages Vgas (Zhang et al. 2019a) Combining ab initio and similarity-based method for predicting viral genes High precision and recall rate Accuracy needs further improvement Taxonomic assignment of the virome vConTACT (Eloe-Fadrosh, 2019) Classification of prokaryotic virome based on the gene sharing network Universal, scalable and automated Not for short fragments, the singleton or outlier sequences; only tested for phages GRAViTy (Eloe-Fadrosh, 2019) Classification of eukaryotic viruses at the family level within each Baltimore group Concise and clear Not for short contigs; only for eukaryotic viruses Evolution of the virome Analyzed the origins and evolution of the RNA virome (Wolf et al. 2018) Reconstruct the RNA virus evolution using the RdRp protein, and reveal extensive gene module exchange and horizontal virus transfer among diverse viruses A far more complete reconstruction of the evolution of RNA viruses than those in previous studies Only for RNA viruses Host prediction of phages PHP (Lu et al. 2021) A Gaussian model for host prediction of prokaryotic viruses in metagenomics Accurate, fast and user-friendly Accuracy needs further improvement Virus-host interactions P-HIPSTer (Lasso et al. 2019) Prediction of PPIs between 1,001 human-infecting viruses and human based on structure information Comprehensive and accurate The codes are not available Predict the receptorome of human viruses (Zhang et al. 2020b) Predicting the receptorome of the human-infecting virome based on the unique features of mammalian virus receptors Fast and comprehensive Accuracy needs further improvement; unable to predict virus-receptor interactions directly Virus culturomics KOMODO (Oberhardt et al. 2015) A platform for recommending microbial media Data-rich, user-friendly and relatively accurate Only suitable for bacteria and archaea Association of virome and human health MicroPro (Zhu et al. 2019) A data analysis pipeline for analysis of the association between the microbes in human body and the complex diseases Combining both known and unknown microbial organisms Did not consider the complex interactions between microbes HumanVirusFinder (Zhang et al. 2019b) A machine learning model for identification of human-infecting viruses from viral metagenomic data Fast, easy to use, suitable for genomes, contigs and reads Limited data used in training the models Table 1. Illustration of computational methods or case studies in computational viromics.
Figure 1 个
Table 1 个