











-
In 1999, we isolated a strain of virus with high hemagglutinin titer from the lung of a dead common cotton-eared marmoset during a respiratory-disease outbreak in an animal laboratory. In the epidemic, almost all the marmosets became ill, and about 1/3 died. Virological and morphological analysis and sequence determination of part of the HN gene indicated that the virus responsible for the outbreak belonged to the family Paramyxoviridae, and so it was temporarily designated paramyxovirus Tianjin strain. In our previous work, a fragment of 375 bp was amplified from viral RNA by RT-PCR and sequenced, then sequence similarity searches were conducted using the BLAST service at the National Center for Biotechnology Information (NCBI). The results showed that the fragment had highest similarity with the part of the HN gene of Sendai virus (SeV) (10).
SeV is a member of the Respirovirus genus in the Paramyxovirinae subfamily. It usually causes out-breaks of lethal pneumonia in mouse colonies, the natural host, but is thought to be nonpathogenic in humans (7). During the epidemic, we found that the experimental mice bred in the same animal laboratory with marmosets had never suffered from the respi-ratory disease. Moreover, we failed to detect anti-bodies against the Tianjin strain in laboratory mice using the ELISA test. We postulate that this strain may be a variant of SeV whose host changed from rodents to marmoset.
In order to understand the genomic structure and taxonomic position of the strain, its complete genome was sequenced and analyzed. Characterization of genome and phylogenetic analysis with other members of the family Paramyxoviridae demonstrated that the Tianjin strain should be assigned to the genus Respirovirus within the subfamily Paramyxovirinae, and was most closely related to SeV (4, 5, 11). The genome of Tianjin strain consists of six structural genes in the order 3'-NP-P/C-M-F-HN-L-5', coding for the nucleocapsid (NP), phosphoprotein (P), matrix (M), fusion (F), hemagglutinin-neuraminidase (HN), and large (L) protein, respectively. In the present paper, the amino acid sequences of six structural proteins of Tianjin strain were analyzed by using the bioinformatics methods. Our findings may provide a useful basis for the vaccine studies.
HTML
-
The full-length genome sequence of Tianjin strain has been deposited in GenBank under accession number EF679198. Accession numbers for other members of the Paramyxoviridae sequences used in this study were as follows: Avian paramyxovirus type 6 (APMV6), NC_003043; Bovine parainfluenza virus 3 (bPIV3), NC_002161; Bovine respiratory syncytial virus (bRSV), NC_001989; Canine distemper virus (CDV), NC_001921; Dolphin morbillivirus (DMV), NC_005283; Fer-de-lance virus (FDLV), NC_005084; Hendra virus (HeV), NC_001906; Human parain-fluenza virus 1 (hPIV1), NC_003461; hPIV2, NC_ 003443; hPIV3, NC_001796; Human respiratory syncytial virus (hRSV), NC_001781; Measles virus (MeV), NC_001498; Menangle virus (MenV), NC_ 007620; Mumps virus (MuV), NC_002200; Newcastle disease virus (NDV), NC_002617; Nipah virus (NiV), NC_002728; Peste-des-petits-ruminants virus (PPRV), NC_006383; Rinderpest virus (RPV), NC_006296; Sendai virus Hamamatsu strain, AB039658; Simian virus 5 (SV5), NC_006430; Tioman virus (TiV), NC_ 004074; Tupaia paramyxovirus (TPMV), NC_002199; J virus (J-V), NC_007454; Avian metapneumovirus (AMPV), NC_007652; Human metapneumovirus (HMPV), NC_004148.
Accession numbers for different strains of SeV used in this study were as follows: Ohita M1, AB005795/ NC_001552; Ohita MVC11, AB005796; Hamamatsu E0, AB039658; Hamamatsu E30cl2, AB065186; Hamamatsu E15cl2, AB065187; Hamamatsu E50cl9, AB065188; Hamamatsu E30M15cl5, AB065189; Pi, AB195967; Nagoya, AB195968; BB1, DQ219803; Z, M30202; F1-R, M30203; mutant ts-f1, M30204; mutant T-5 revertant, M69046.
-
The raw data files were edited using the EditSeq program in the DNAStar software package (Lasergene). Multiple alignments of deduced amino acid sequences between Tianjin strain and 14 known SeVs were performed with the DNAStar MegAlign program (version 5.01) using the ClustalW method. Identity values were determined from the generated align-ments. Phylogenetic analyses were performed using Clustal X1.83 on the basis of amino acid sequence alignments of the NP, P, M, F, HN and L proteins. The bootstrap values were calculated for 1000 repli-cates. Phylogeny trees were drawn using TreeView, version 1.6.6.
GenBank accession numbers
Sequence analysis
-
The deduced amino acid sequences of the full-length NP, P, M, F, HN, and L genes of the Tianjin strain were used to infer phylogenetic relationships with other Paramyxoviridae. Representative trees illus-trating the relationships observed are shown in Fig. 1. In the P and L protein trees, the genera Rubulavirus, Morbillivirus, Respirovirus, Henipavirus, Avulavirus, Pneumovirus and Metapneumovirus separated into distinct clusters. This result is consistent with the most recent taxonomy approved by the ICTV executive committee. Moreover, the Tianjin strain formed a single cluster with SeV, hPIV-1, hPIV-3 and bPIV-3 and was more closely related to SeV than to any other virus in the same genus. The NP, M, F and HN protein trees were similar in showing that the Tianjin strain was positioned in the Respirovirus genus and was most closely related to SeV (data not shown). Furthermore, phylogenetic analysis based on 6 structural proteins of the Tianjin strain and 14 strains of SeV clearly showed that the viruses were roughly divided into four phylogenetic clusters, the first containing Ohita M1, MVC11and Hamamatsu E0, E30cl2, E15cl2, E50cl9 and E30M15cl5, the second containing Pi, Nagoya, Z, F1-R, mutant ts-f1 and mutant T-5 revertant, and the third represented by BB1 strain. The Tianjin strain formed an independent branch (P, M and L protein trees shown in Fig. 2). This result suggested that Tianjin strain was most likely a new genotype of SeV. However, trees of the different proteins were not identical. The NP, P, F, HN and L protein trees were similar in showing Tianjin strain was most closely related to BB1 strain, whereas in the M protein trees, Tianjin strain was more closely related to the branch represented by Ohita and Hamamatsu strain than to BB1 strain. Similarity comparison of M proteins between the Tianjin strain and known SeVs also demonstrated that the Tianjin strain shared higher identity value with Ohita and Hamamatsu strain than with the BB1 strain (Table 1). This suggests a somewhat different evolutionary pathway of the Tianjin strain M protein compared to the NP, P, F, HN, and L proteins.

Figure 1. The Neighbor-Joining trees based on the complete P (A) and L protein (B) sequences of Tianjin strain (see arrow) and other Paramyxoviridae. The numbers on the nodes represent bootstrap values (for 1000 replications).

Figure 2. The Neighbor-Joining trees based on the complete P (A), M (B) and L protein (C) sequences of Tianjin strain (see arrow) and 14 known SeVs. The numbers on the nodes represent bootstrap values (for 1000 replications).

Table 1. Similarities comparisons of amino acid sequences of some proteins between Tianjin strain and 14 known SeVs (%)
-
Similarities comparisons of Tianjin strain protein sequences with those of 14 known SeVs clearly showed that L protein was relatively well conserved, sharing 96% to 98% amino acid identity with the known SeVs, followed by the M (93.1%~97.1%), NP (92.2%~96.0%), F (90.4%~95.4%), and HN proteins (89.4%~95.0%). The Tianjin strain P protein had relatively low levels of amino acid identity with the P proteins of SeVs, ranging from 78.7% to 91.9% (Table 1). This pattern was consistent with previous descriptions in which the P protein of SeV was poorly conserved.
-
When the alignments were carried out on the protein sequences of Tianjin strain and analogous proteins of 14 known SeVs, 110 unique amino acid substitutions were found, 15 in NP, 29 in P, 6 in M, 13 in F, 18 in HN, and 29 in L (see Fig. 3, which shows the alignment of M proteins). Among these unique amino acid substitutions, there were totally 54 conservative substitutions, 9 in NP, 8 in P, 2 in M, 10 in F, 10 in HN, and 15 in L. The presence of these unique amino acid substitutions suggested that the Tianjin strain maybe had a significant difference in biological, pathological, immunological, or epidemio-logical characteristics from the known SeVs. To establish the relationship between the substitutions and viral biological properties or pathogenesis, gene-ration of recombinant SeV carrying the mutation by reverse genetic technology would be necessary.
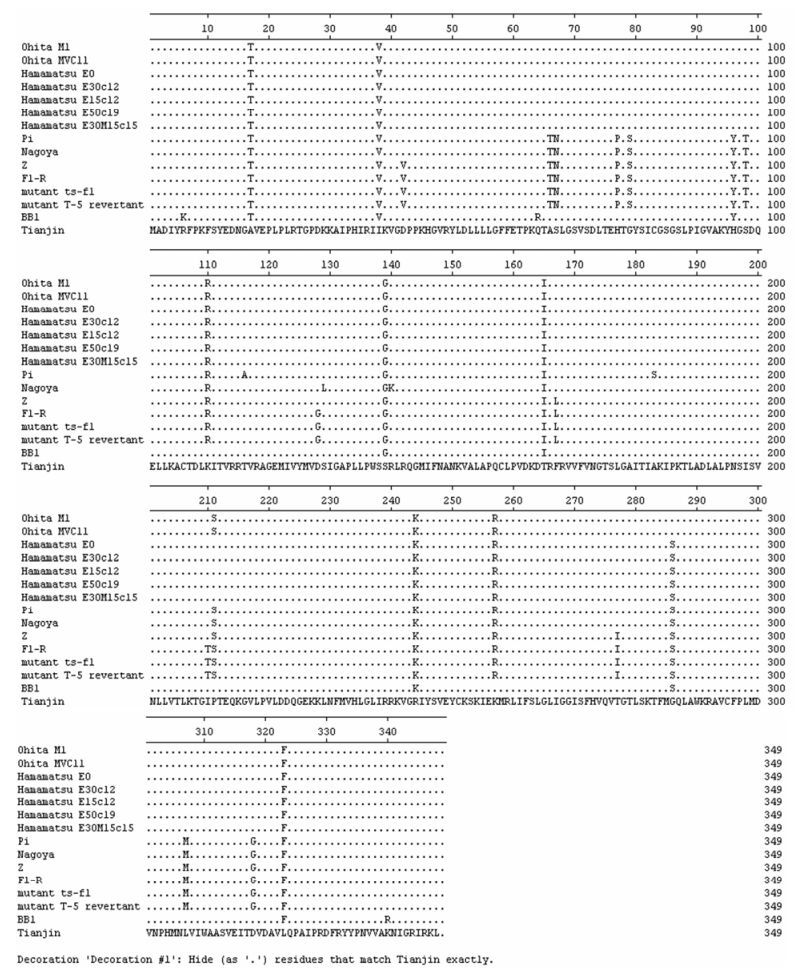
Figure 3. Sequence alignment of M proteins among Tianjin strain and the known SeVs. The arrows indicate the conservative substitutions in M protein sequence of Tianjin strain.
Phylogenetic analysis
Similarities comparisons
Distribution of amino acid substitutions
-
A paramyxovirus designated paramyxovirus Tianjin strain was isolated from the lung of a deceased common cotton-eared marmoset. Its genome consists of six structural genes in the order 3'-NP-P/C-M-F-HN-L-5', coding for NP, P, M, F, HN and L protein, respectively. The Tianjin strain NP protein is 524-amino-acid (aa) in length. We found that C-terminal (20%) of the NP protein was poorly conserved, while the amino-terminal (80%) of the protein was relatively well conserved. Within the conserved domain of the NP protein, the Y260 and F324 residues that have been reported to be conserved in all Paramyxovirinae are present in the Tianjin strain. These amino acids are important for RNA binding and NP-NP protein self-assembly respectively (6). In the Tianjin strain F324 also marks the start of a larger conserved motif found near the middle of all paramyxovirus NP proteins, F-X4-Y-X4-SYAMG (where X represents any amino acid residue).
In SeV, aa residues 33 to 41 of the P protein were demonstrated to be essential for genome replication (1). This 9-aa domain is also found in the putative 568-aa Tianjin strain P protein, suggesting it could also play an important role in genome replication of Tianjin strain. In addition, aa residues 506 to 509, aa 524 to 526 and aa 560 to 562 in the C-terminal of the SeV P protein have been identified to be responsible for binding nucleocapsid (assembled NP) by site-directed mutagenesis (12). The aa residues in these domains of the Tianjin strain P protein are identical to those of SeV, indicating Tianjin strain perhaps has RNA polymerase activity.
The Tianjin strain M protein is 348 amino acids in length. Like the M proteins of SeV, the Tianjin strain M protein contains five cysteine residues, at positions 83, 106, 158, 251, and 295. In fact, cysteine residues are well conserved in a wide variety of paramyxovirus M proteins suggesting that the functions of the M proteins are important. In SeV, it has been proved that a single-point mutation at a cysteine residue of the M protein affects virus morphology and nucleocapsid incorporation, indicating direct involvement of the M protein in SeV assembly (9).
The Tianjin strain F ORF encodes a 565-aa protein that was found be closest in sequence to that of SeV by BLAST analysis. Hydrophobicity and transme-mbrane analysis confirmed that the Tianjin strain F protein had features of a type I integral membrane protein. These features included a putative signal sequence at the N-terminal 25 aa and a transme-mbrane domain of approximately 24 aa near the carboxy terminus (aa 500 to 523), followed by a carboxy-terminal hydrophilic tail of 42 aa. The predi-cted cleavage site of the Tianjin strain F protein, GVPQAR at aa 111 to 116, is monobasic and does not conform to the consensus motif for cleavage by furin, R-X-K/R-R (2), conserved in the majority of the Paramyxovirinae (morbilliviruses, rubulaviruses, avula-viruses, pneumoviruses, and some respiroviruses). It is most similar to the cleavage sites of wild-type SeV (DVPQSR). It has been shown that wild-type SeV causes a pneumotropic infection in rodents, while F1-R, a pantropic variant of SeV, causes a systemic infection (13). The primary determinant of the pantro-pism is the exchange (Ser to Pro) residue 115 next to the cleavage site of the F protein that results in the enhanced cleavability of the F protein. The aa residue 115 of the Tianjin strain F protein changes from Ser to Ala, the role of which remains unknown.
The Tianjin strain HN ORF encodes a 575-aa protein. Analysis of hydrophobic and transmembrane domains indicated that the Tianjin strain HN protein had features of a type II membrane protein. There was a hydrophobic 25-aa putative transmembrane domain near the amino terminus (aa 36 to 60) following an amino-terminal cytoplasmic tail of 35 aa. In addition, the Tianjin strain HN protein contains the sialic acid binding site motif N-R-K-S-C-S (aa 254 to 259) that is conserved in respiroviruses and rubulaviruses but is not present in morbilliviruses, henipaviruses, or MenV (3). This finding suggests the probability that Tianjin strain uses a sialic acid-containing cellular receptor similar to respiroviruses and rubulaviruses.
The L gene of Tianjin strain is 6 690 nt long and has a single large ORF starting with an AUG codon in strong initiation context. This ORF encodes a 2 229-aa protein. Alignment with the L gene of different strains of SeV demonstrates that a single nucleotide substi-tution (U to G) occurs at nt 6 685 of the Tianjin strain L gene resulting in the stop codon UAA to GAA, leading to the extended L protein. It is interesting that SeV BB1 strain has the same mutation site. Thus the Tianjin strain L protein is identical in length to that of SeV BB1 strain, which is one amino acid (Glu) more than other known SeVs L proteins in length.
In order to demonstrate the taxonomic position of Tianjin strain, phylogenetic analysis were carried out on protein sequences among Tianjin strain and 25 paramyxoviruses. All of the phylogenetic trees clearly showed that Tianjin strain belonged to the genus Respirovirus and was most closely related to SeV. To clarify the relationship between the Tianjin strain and SeV, phylogenetic analysis based on 6 structural proteins were performed with the Tianjin strain and 14 known SeVs. The results showed that the viruses were divided into four evolutionary lineages and the Tianjin strain possessed a novel evolutionary lineage. De et al reported that sequenced SeV strains could be divided into two evolutionary lineages by nucleotide sequence comparison. Conventional laboratory strains such as Z and Fushimi were in one lineage, and those isolated in the 1970s in Japan such as Hamamatsu and Ohita were in the other lineage (8, 14). In addition, Yang et al reported the complete sequence of SeV BB1 strain and posed that the BB1 strain was not like the two evolutional lineages described above and belonged to a third different genetic group (15). Our result verifies the previous reports and suggests that Tianjin strain is most likely a new genotype of SeV, which might provide an important basis for the classification of SeV on the molecular level.
Interestingly, all of the protein trees and identity values except M protein showed that the Tianjin strain was more closely related to the BB1 strain than any other virus. The BB1 strain was isolated from mice in Moscow, but sequenced and submitted by Institute of Viral Disease Control and Prevention in Beijing, China. Moreover, among all the SeV strains sequenced to date, only BB1 strain is identical to Tianjin strain in the L gene length. We postulate that the high similarity between them is presumably due to the effect of region factor on viral evolution.

 DownLoad:
DownLoad: