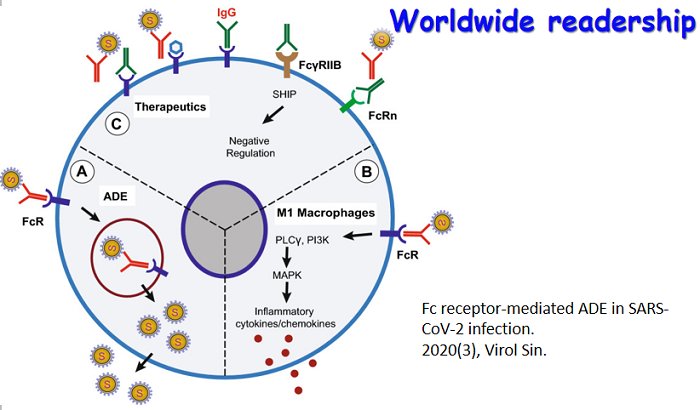
Guaico Culex virus (GCXV) is a newly identified segmented Jingmenvirus found inCulex spp. mosquitoes in Central and South America. The genome of GCXV consists of four or five single-stranded positive RNA segments. However, the infection kinetics and transmission capability of GCXV in mosquitoes remain unknown. In this tissue, Chen et al. utilized reverse genetics to rescue two GCXVs (4S and 5S) that contained four and five RNA segments, respectively, in C6/36 cells. Both GCXVs exhibited comparable replication kinetics in C6/36 cells. Notably, both GCXVs showed limited replication in Cx. quinquefasciatus after oral infection. Additionally GCXV RNAs were detected in Cx. quinquefasciatus eggs during the first and second gonotrophic cycles after oral infection. The cover image illustrates the limited infectivity and dissemination of GCXVs in Cx. quinquefasciatus mosquitoes, highlighting the importance of continuous surveillance of mosquito-borne viruses (Kindly designed and provided by Prof. Fei Wang, Yong-Qiang Deng and Cheng-Feng Qin). See page 228–234 for details.
The persistent epidemic of human mpox, caused by mpox virus (MPXV), raises concerns about the future spread of MPXV and other poxviruses. MPXV is a typical zoonotic virus which can infect human and cause smallpox-like symptoms. MPXV belongs to the Poxviridae family, which has a relatively broad host range from arthropods to vertebrates. Cross-species transmission of poxviruses among different hosts has been frequently reported and resulted in numerous epidemics. Poxviruses have a complex linear double-strand DNA genome that encodes hundreds of proteins. Genes related to the host range of poxvirus are called host range genes (HRGs). This review briefly introduces the taxonomy, phylogeny and hosts of poxviruses, and then comprehensively summarizes the current knowledge about the cross-species transmission of poxviruses. In particular, the HRGs of poxvirus are described and their impacts on viral host range are discussed in depth. We hope that this review will provide a comprehensive perspective about the current progress of researches on cross-species transmission and HRG variation of poxviruses, serving as a valuable reference for academic studies and disease control in the future.
Haemaphysalis longicornis ticks, commonly found in East Asia, can transmit various pathogenic viruses, including the severe fever with thrombocytopenia syndrome virus (SFTSV) that has caused febrile diseases among humans in Hubei Province. However, understanding of the viromes of H. longicornis was limited, and the prevalence of viruses among H. longicornis ticks in Hubei was not well clarified. This study investigates the viromes of both engorged (fed) and free (unfed) H. longicornis ticks across three mountainous regions in Hubei Province from 2019 to 2020. RNA-sequencing analysis identified viral sequences that were related to 39 reference viruses belonging to unclassified viruses and seven RNA viral families, namely Chuviridae, Nairoviridae, Orthomyxoviridae, Parvoviridae, Phenuiviridae, Rhabdoviridae, and Totiviridae. Viral abundance and diversity in these ticks were analysed, and phylogenetic characteristics of the Henan tick virus (HNTV), Dabieshan tick virus (DBSTV), Okutama tick virus (OKTV), and Jingmen tick virus (JMTV) were elucidated based on their full genomic sequences. Prevalence analysis demonstrated that DBSTV was the most common virus found in individual H. longicornis ticks (12.59%), followed by HNTV (0.35%), whereas JMTV and OKTV were not detected. These results improve our understanding of H. longicornis tick viromes in central China and highlight the role of tick feeding status and geography in shaping the viral community. The findings of new viral strains and their potential impact on public health raise the need to strengthen surveillance efforts for comprehensively assessing their spillover potentials.
Swine are regarded as “intermediate hosts” or “mixing vessels” of influenza viruses, capable of generating strains with pandemic potential. From 2020 to 2021, we conducted surveillance on swine H1N2 influenza (swH1N2) viruses in swine farms located in Guangdong, Yunnan, and Guizhou provinces in southern China, as well as Henan and Shandong provinces in northern China. We systematically analyzed the evolution and pathogenicity of swH1N2 isolates, and characterized their replication and transmission abilities. The isolated viruses are quadruple reassortant H1N2 viruses containing genes from pdm/09 H1N1 (PB2, PB1, PA and NP genes), triple-reassortant swine (NS gene), Eurasian Avian-like (HA and M genes), and recent human H3N2 (NA gene) lineages. The NA, PB2, and NP of SW/188/20 and SW/198/20 show high gene similarities to A/Guangdong/Yue Fang277/2017 (H3N2). The HA gene of swH1N2 exhibits a high evolutionary rate. The five swH1N2 isolates replicate efficiently in human, canine, and swine cells, as well as in the turbinate, trachea, and lungs of mice. A/swine/Shandong/198/2020 strain efficiently replicates in the respiratory tract of pigs and effectively transmitted among them. Collectively, these current swH1N2 viruses possess zoonotic potential, highlighting the need for strengthened surveillance of swH1N2 viruses.
The SARS-CoV-2 Omicron variants are notorious for their transmissibility, but little is known about their subgenomic RNA (sgRNA) expression. This study applied RNA-seq to delineate the quantitative and qualitative profiles of canonical sgRNA of 118 respiratory samples collected from patients infected with Omicron BA.2 and compared with 338 patients infected with non-variant of concern (non-VOC)-D614G. A unique characteristic profile depicted by the relative abundance of 9 canonical sgRNAs was reproduced by both BA.2 and non-VOC-D614G regardless of host gender, age and presence of pneumonia. Remarkably, such profile was lost in samples with low viral load, suggesting a potential application of sgRNA pattern to indicate viral activity of individual patient at a specific time point. A characteristic qualitative profile of canonical sgRNAs was also reproduced by both BA.2 and non-VOC-D614G. The presence of a full set of canonical sgRNAs carried a coherent correlation with crude viral load (AUC = 0.91, 95% CI 0.88–0.94), and sgRNA ORF7b was identified to be the best surrogate marker allowing feasible routine application in characterizing the infection status of individual patient. Further potentials in using sgRNA as a target for vaccine and antiviral development are worth pursuing.
Guaico Culex virus (GCXV) is a newly identified segmented Jingmenvirus from Culex spp. mosquitoes in Central and South America. The genome of GCXV is composed of four or five single-stranded positive RNA segments. However, the infection kinetics and transmission capability of GCXV in mosquitoes remain unknown. In this study, we used reverse genetics to rescue two GCXVs (4S and 5S) that contained four and five RNA segments, respectively, in C6/36 cells. Further in vitro characterization revealed that the two GCXVs exhibited comparable replication kinetics, protein expression and viral titers. Importantly, GCXV RNAs were detected in the bodies, salivary glands, midguts and ovaries of Culex quinquefasciatus at 4–10 days after oral infection. In addition, two GCXVs can colonize Cx. quinquefasciatus eggs, resulting in positive rates of 15%–35% for the second gonotrophic cycle. In conclusion, our results demonstrated that GCXVs with four or five RNA segments can be detected in Cx. quinquefasciatus eggs during the first and second gonotrophic cycles after oral infection.
Inclusion bodies (IBs) of respiratory syncytial virus (RSV) are formed by liquid-liquid phase separation (LLPS) and contain internal structures termed “IB-associated granules” (IBAGs), where anti-termination factor M2-1 and viral mRNAs are concentrated. However, the mechanism of IBAG formation and the physiological function of IBAGs are unclear. Here, we found that the internal structures of RSV IBs are actual M2-1-free viral messenger ribonucleoprotein (mRNP) condensates formed by secondary LLPS. Mechanistically, the RSV nucleoprotein (N) and M2-1 interact with and recruit PABP to IBs, promoting PABP to bind viral mRNAs transcribed in IBs by RNA-recognition motif and drive secondary phase separation. Furthermore, PABP-eIF4G1 interaction regulates viral mRNP condensate composition, thereby recruiting specific translation initiation factors (eIF4G1, eIF4E, eIF4A, eIF4B and eIF4H) into the secondary condensed phase to activate viral mRNAs for ribosomal recruitment. Our study proposes a novel LLPS-regulated translation mechanism during viral infection and a novel antiviral strategy via targeting on secondary condensed phase.
Viral encephalitis continues to be a significant public health concern. In our previous study, we discovered a lower expression of antiviral factors, such as IFN-β, STING and IFI16, in the brain tissues of patients with Rasmussen's encephalitis (RE), a rare chronic neurological disorder often occurred in children, characterized by unihemispheric brain atrophy. Furthermore, a higher cumulative viral score of human herpes viruses (HHVs) was also found to have a significant positive correlation with the unihemispheric atrophy in RE. Type I IFNs (IFN-I) signaling is essential for innate anti-infection response by binding to IFN-α/β receptor (IFNAR). In this study, we infected WT mice and IFNAR-deficient A6 mice with herpes simplex virus 1 (HSV-1) via periocular injection to investigate the relationship between IFN-I signaling and HHVs-induced brain lesions. While all mice exhibited typical viral encephalitis lesions in their brains, HSV-induced epilepsy was only observed in A6 mice. The gene expression matrix, functional enrichment analysis and protein-protein interaction network revealed four gene models that were positively related with HSV-induced epilepsy. Additionally, ten key genes with the highest scores were identified. Taken together, these findings indicate that intact IFN-I signaling can effectively limit HHVs induced neural symptoms and brain lesions, thereby confirming the positive correlation between IFN-I signaling repression and brain atrophy in RE and other HHVs encephalitis.
Porcine reproductive and respiratory syndrome virus (PRRSV) is a major economically devastating pathogen that has evolved various strategies to evade innate immunity. Downregulation of antiviral interferon largely promotes PRRSV immunoevasion by utilizing cytoplasmic melanoma differentiation-associated gene 5 (MDA5), a receptor that senses viral RNA. In this study, the downregulated transcription and expression levels of porcine MDA5 in PRRSV infection were observed, and the detailed mechanisms were explored. We found that the interaction between P62 and MDA5 is enhanced due to two factors: the phosphorylation modification of the autophagic receptor P62 by the upregulated kinase CK2α and the K63 ubiquitination of porcine MDA5 catalyzed by the E3 ubiquitinase TRIM21 in PRRSV-infected cells. As a result of these modifications, the classic P62-mediated autophagy is triggered. Additionally, porcine MDA5 interacts with the chaperonin containing TCP1 subunit 2 (CCT2), which is enhanced by PRRSV nsp3. This interaction promotes the aggregate formation and autophagic clearance of MDA5-CCT2-nsp3 independently of ubiquitination. In summary, enhanced MDA5 degradation occurs in PRRSV infection via two autophagic pathways: the binding of MDA5 with the autophagy receptor P62 and the aggrephagy receptor CCT2, leading to intense innate immune suppression. The research reveals a novel mechanism of immune evasion in PRRSV infection and provides fundamental insights for the development of new vaccines or therapeutic strategies.
Influenza A virus (IAV) binds sialic acid receptors on the cell surface to enter the host cells, which is the key step in initiating infection, transmission and pathogenesis. Understanding the factors that contribute to the highly efficient entry of IAV into human cells will help elucidate the mechanism of viral entry and pathogenicity, and provide new targets for intervention. In the present study, we reported a novel membrane protein, C1QTNF5, which binds to the hemagglutinin protein of IAV and promotes IAV infection in vitro and in vivo. We found that the HA1 region of IAV hemagglutinin is critical for the interaction with C1QTNF5 protein, and C1QTNF5 interacts with hemagglutinin mainly through its N-terminus (1–103 aa). In addition, we further demonstrated that overexpression of C1QTNF5 promotes IAV entry, while blocking the interaction between C1QTNF5 and IAV hemagglutinin greatly inhibits viral entry. However, C1QTNF5 does not function as a receptor to mediate IAV infection in sialic acid-deficient CHO-Lec2 cells, but promotes IAV to attach to these cells, suggesting that C1QTNF5 is an important attachment factor for IAV. This work reveals C1QTNF5 as a novel IAV attachment factor and provides a new perspective for antiviral strategies.
Coxsackievirus B3 (CVB3) is the pathogen causing hand, foot and mouth disease (HFMD), which manifests across a spectrum of clinical severity from mild to severe. However, CVB3-infected mouse models mainly demonstrate viral myocarditis and pancreatitis, failing to replicate human HFMD symptoms. Although several enteroviruses have been evaluated in Syrian hamsters and rhesus monkeys, there is no comprehensive data on CVB3. In this study, we have first tested the susceptibility of Syrian hamsters to CVB3 infection via different routes. The results showed that Syrian hamsters were successfully infected with CVB3 by intraperitoneal injection or nasal drip, leading to nasopharyngeal colonization, acute severe pathological injury, and typical HFMD symptoms. Notably, the nasal drip group exhibited a longer viral excretion cycle and more severe pathological damage. In the subsequent study, rhesus monkeys infected with CVB3 through nasal drips also presented signs of HFMD symptoms, viral excretion, serum antibody conversion, viral nucleic acids and antigens, and the specific organ damages, particularly in the heart. Surprisingly, there were no significant differences in myocardial enzyme levels, and the clinical symptoms resembled those often associated with common, mild infections. In summary, the study successfully developed severe Syrian hamsters and mild rhesus monkey models for CVB3-induced HFMD. These models could serve as a basis for understanding the disease pathogenesis, conducting pre-trial prevention and evaluation, and implementing post-exposure intervention.
Hand, foot, and mouth disease (HFMD) is a common pediatric illness mainly caused by enteroviruses, which are important human pathogens. Currently, there are no available antiviral agents for the therapy of enterovirus infection. In this study, an excellent high-content antiviral screening system utilizing the EV-A71-eGFP reporter virus was developed. Using this screening system, we screened a drug library containing 1042 natural compounds to identify potential EV-A71 inhibitors. Fangchinoline (FAN), a bis-benzylisoquinoline alkaloid, exhibits potential inhibitory effects against various enteroviruses that cause HFMD, such as EV-A71, CV-A10, CV-B3 and CV-A16. Further investigations revealed that FAN targets the early stage of the enterovirus life cycle. Through the selection of FAN-resistant EV-A71 viruses, we demonstrated that the VP1 protein could be a potential target of FAN, as two mutations in VP1 (E145G and V258I) resulted in viral resistance to FAN. Our research suggests that FAN is an efficient inhibitor of EV-A71 and has the potential to be a broad-spectrum antiviral drug against human enteroviruses.
SARS-CoV-2 infection-induced hyper-inflammation is a key pathogenic factor of COVID-19. Our research, along with others', has demonstrated that mast cells (MCs) play a vital role in the initiation of hyper-inflammation caused by SARS-CoV-2. In previous study, we observed that SARS-CoV-2 infection induced the accumulation of MCs in the peri-bronchus and bronchioalveolar-duct junction in humanized mice. Additionally, we found that MC degranulation triggered by the spike protein resulted in inflammation in alveolar epithelial cells and capillary endothelial cells, leading to subsequent lung injury. The trachea and bronchus are the routes for SARS-CoV-2 transmission after virus inhalation, and inflammation in these regions could promote viral spread. MCs are widely distributed throughout the respiratory tract. Thus, in this study, we investigated the role of MCs and their degranulation in the development of inflammation in tracheal-bronchial epithelium. Histological analyses showed the accumulation and degranulation of MCs in the peri-trachea of humanized mice infected with SARS-CoV-2. MC degranulation caused lesions in trachea, and the formation of papillary hyperplasia was observed. Through transcriptome analysis in bronchial epithelial cells, we found that MC degranulation significantly altered multiple cellular signaling, particularly, leading to upregulated immune responses and inflammation. The administration of ebastine or loratadine effectively suppressed the induction of inflammatory factors in bronchial epithelial cells and alleviated tracheal injury in mice. Taken together, our findings confirm the essential role of MC degranulation in SARS-CoV-2-induced hyper-inflammation and the subsequent tissue lesions. Furthermore, our results support the use of ebastine or loratadine to inhibit SARS-CoV-2-triggered degranulation, thereby preventing tissue damage caused by hyper-inflammation.
Naturally occurred precore (PC, G1896A) and/or basal core promoter (BCP, A1762T/G1764A) mutations are prevalent in chronic HBV-infected patients, especially those under HBeAg-negative status. However, the replicative capacity of HBV with PC/BCP mutations remains ambiguous. Herein, meta-analysis showed that, only under HBeAg-negative status, the serum HBV DNA load in patients with PC mutation was 7.41-fold higher than those without the mutation. Both PC mutation alone and BCP + PC mutations promoted HBV replication in cell and hydrodynamic injection mouse models. In human hepatocyte chimeric mouse model, BCP + PC mutations led to elevated replicative capacity and intrahepatic core protein accumulation. Mechanistically, preC RNA harboring PC mutation could serve as mRNA to express core and P proteins, and such pgRNA-like function favored the maintenance of cccDNA pool under HBeAg-negative status. Additionally, BCP + PC mutations induced more extensive and severe human hepatocyte damage as well as activated endoplasmic reticulum stress and TNF signaling pathway in livers of chimeric mice. This study indicates that HBeAg-negative patients should be monitored on HBV mutations regularly and are expected to receive early antiviral treatment to prevent disease progression.
Highlights 1. Phospholipid-binding abilities of mpox virus A7 protein and its truncations are investigated. 2. The structures of the N-terminal truncations of A7 protein (A7N121 and A7N137) are determined. 3. Conformational changes of the conserved linking helix in A7 are illustrated. 4. A structural model of the full-length A7 protein is proposed.
Highlights 1. We reported the first MPXV strain hMpxV/China/SZ-SZTH42/2023 isolated in southern China. 2. The isolate SZTH42 belongs to C.1 lineage of clade IIb, representing the currently prevalent IIb branch strain worldwide. 3. This study provides key resources and technical platforms for further research as well as antiviral drugs and vaccines development.
Highlights 1. The expression level of TRIM21 in patients is negatively correlated with the replication and integration of HBV. 2. TRIM21 was found to trigger non-proteolytic ubiquitination of X protein of HBV. 3. This study proposes that the PRYSPRY and RING domains in TRIM21 dimer can form a docking conformation for HBx binding. 4. TRIM21-mediated HBx ubiquitination disrupts the DDB1 recruitment to HBx and stabilize Smc6.
Highlights 1. Single-nucleotide polymorphisms in critical patients with COVID-19 conducted worldwide were validated in Chinese population. 2. Variants in DPP9 and near RNU2-47P and TYRP1 showed the strongest associated signals with COVID-19 severity in Chinese population. 3. Rs1886814 in FOXP4 was correlated with COVID-19 severity in Chinese and other populations.
Due to our negligence, the original version of this article, published online on Mar 14, 2023, contained some mistakes in several Figs. In Fig. 2D, the positions of the bands for protein 3A and 3B were incorrectly shifted. This has been modified in corrected Fig.2 as shown below.