







鉴定HCoVs和SARSr-CoV-2中SARS-CoV-2的RNA条形码片段
新型冠状病毒(SARS-CoV-2)作为引起新型冠状病毒感染(COVID-19)的病原体,不断进化产生新变异体,进而导致全球范围内的疫情复发。先前研究表明,条形码片段可以在具有密切系统发育关系的群体中高效、低成本地识别特定物种。在本研究中,基于物种的遗传演化关系,我们构建并测试了SARS-CoV-2的RNA条形码片段,便于从大规模的病毒样本中(例如HCoVs和SARSr-CoV-2谱系)高效且准确地识别SARS-CoV-2。我们从NCBI和GISAID数据库筛选并整理了1733个HCoVs和SARSr-CoV-2谱系的全基因组核苷酸序列用以构建测试集。通过物种的遗传水平测试,验证了条形码片段识别SARS-CoV-2的准确性和可靠性。随后,基于单核苷酸多态性位点和加权分数值大小,我们截取并筛选了75个位于ORF1ab、S、E、ORF7a和N编码区的主要和次要物种SARS-CoV-2特异性条形码片段。经测试,这些片段在鉴定性能上的召回率(接近100%),核苷酸水平上的特异性(接近30%)和精度(100%)表现优秀。最终,以上片段以一维和二维组合条形码的形式实现可视化,并存储至在线数据库(http://virusbarcodedatabase.top/)中。条形码技术鉴定SARS-CoV-2的成功应用不仅为涉及完整基因组序列多态性分析的研究提供有价值的见解。此外,这种高效且具有成本效益的鉴定方法也为未来病毒监测相关研究工作提供了一定参考价值。
more >>
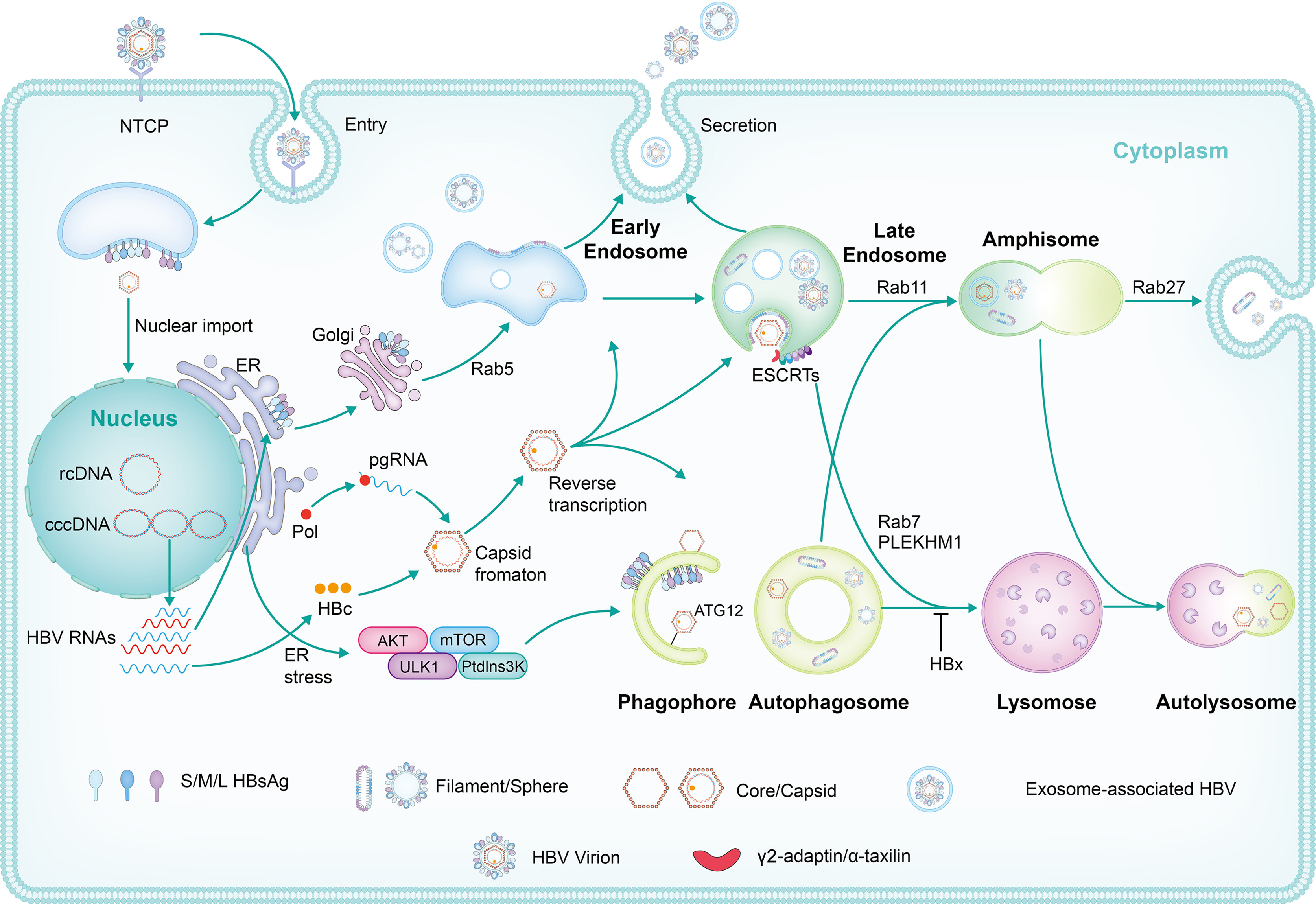
细胞自噬与内体囊泡运输的相互关系及其在乙型肝炎病毒复制和释放中的作用
乙型肝炎病毒(HBV)产生并释放多种类型病毒颗粒,包括病毒体、空包膜、裸衣壳。最近的研究表明,HBV利用不同的细胞内膜运输途径来组装和释放病毒和亚病毒颗粒,包括内体囊泡运输和自噬途径。在此,我们总结了关于自噬、内体膜运输、以及两者的联系在HBV复制、组装和释放中的作用和机制的最新进展。
more >>
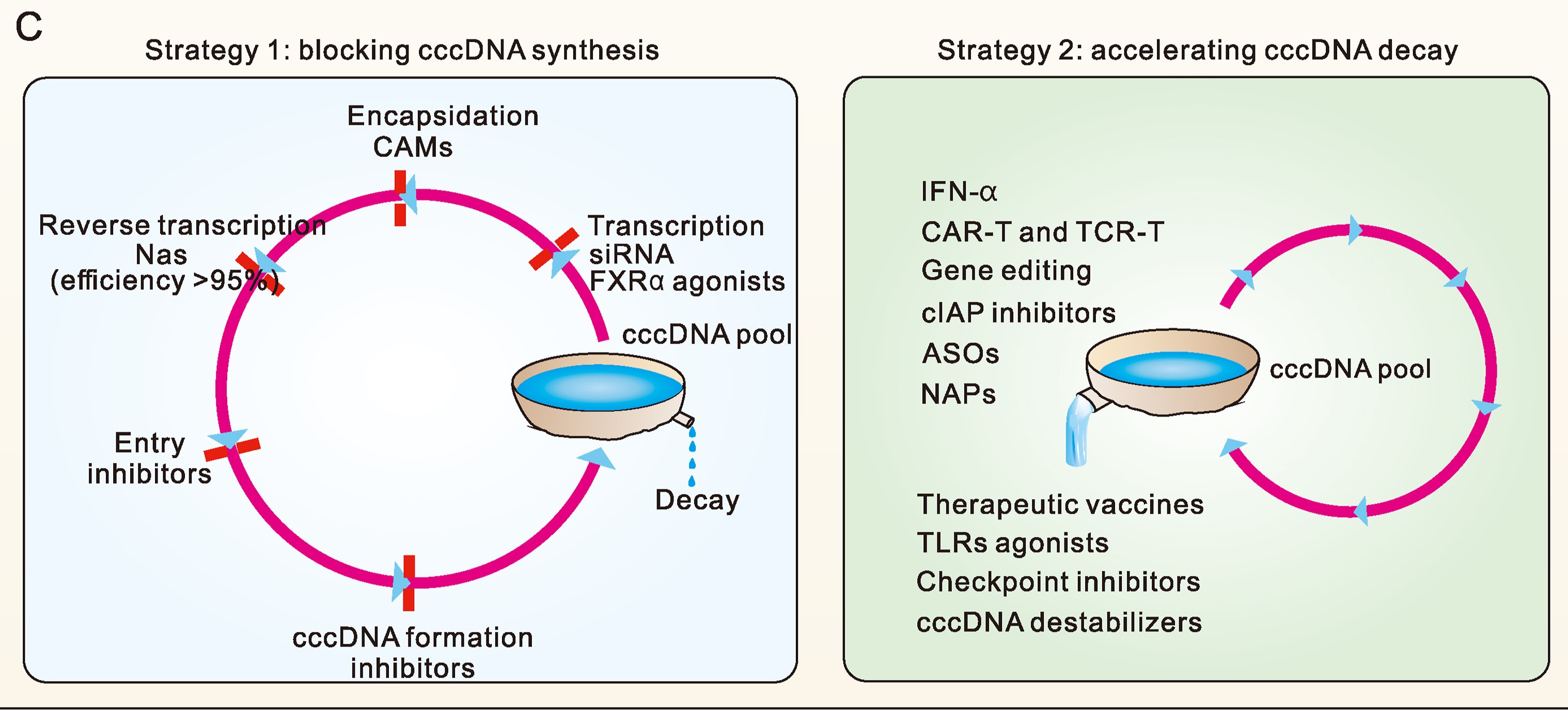
基于cccDNA动力学的乙型肝炎治疗策略分类
现有抗乙肝药物治疗仅能使少数慢性乙型肝炎患者达到功能性治愈。提高功能性治愈率,是当前开发新型抗乙肝药物的主要目标。功能性治愈对应了肝内cccDNA大幅下降乃至清除,因此,抗乙肝治疗过程中cccDNA水平的动态变化是与治愈密切相关的重要问题。我们前期通过分析影响cccDNA动力学的主要因素,初步提出基于cccDNA动力学的乙肝治疗策略分类方式。本文中,我们采用系统思考视角,阐释了乙肝病毒复制循环的本质,分析了各种治疗策略对cccDNA动态平衡的影响,论述了基于cccDNA动力学对治疗策略进行分类的合理性。在此基础上,本文将抗乙肝策略分为两类:阻止cccDNA合成,或者促进cccDNA衰减,并提出,联合这两类策略才能在有限疗程内有效降低cccDNA水平,进一步提高慢乙肝功能性治愈率。
more >>
铁死亡促进日本脑炎病毒引起的神经元损伤和神经炎症
铁死亡是一种新发现的、由铁依赖性脂质过氧化驱动的程序性细胞死亡方式,与多种器官损伤和退行性病变过程有关。尽管研究表明多种细胞死亡方式参与日本脑炎病毒(JEV)感染引起的神经炎症和神经元损伤,但目前尚不清楚铁死亡是否参与其中。在本研究中,我们在体外和体内模型中发现了JEV感染能够引起神经元铁死亡。结果表明,JEV感染通过抑制GSH/GPX4介导的抗氧化系统的功能,并通过促进YAP1/ACSL4介导的脂质过氧化来诱导神经元铁死亡。进一步的分析表明,JEV的C和prM蛋白在此过程中发挥重要作用。此外,本研究通过小鼠感染模型证明使用铁死亡抑制剂能够降低JEV感染小鼠脑中的病毒滴度和炎症反应,并提高感染小鼠的存活率。本研究揭示了铁死亡在JEV致病中的关键作用,并为病毒性脑炎的预防和治疗提供了新的思路。
more >>
PB2 S181磷酸化位点抑制高致病性禽流感病毒在小鼠中的复制和毒力
甲型流感病毒(IAV)仍对公共卫生构成大流行的威胁,每年和在大流行期间造成高死亡率。病毒蛋白的翻译后修饰在调控IAV感染中起着重要作用。基于免疫沉淀(IP)联合质谱(MS)和纯化的病毒联合MS的方法,本研究鉴定出89个磷酸化位点,分布在10个IAV编码的病毒蛋白中,其中包括60个新的磷酸化位点。此外,我们首次提供了PB2也可以被乙酰化的证据,且该位点为K187。值得注意的是,PB2 S181磷酸化位点在IP-MS和纯化病毒联合MS中被重复鉴定到。S181和K187均暴露在PB2蛋白表面,在各种IAV菌株中高度保守,表明它们在IAV生命周期中具有重要意义。生物信息学分析结果显示,S181E/A和K187Q/R模拟突变并没有显著改变PB2蛋白的结构。PB2 S181E突变模拟的持续磷酸化显著降低了病毒对小鼠的毒力,而PB2 K187Q模拟乙酰化略微增强了病毒对小鼠的毒力。在机制上,PB2 S181E显著抑制了病毒的聚合酶活性和病毒复制,显著抑制了PB2蛋白的稳定性和核聚集能力,并显著削弱了IAV诱导的炎症反应。因此,我们的研究进一步丰富了流感病毒蛋白磷酸化和乙酰化位点的数据库,为后续的机制研究奠定了基础。同时,所发现的PB2 S181E模拟磷酸化抗病毒作用可能为后续抗病毒药物的研究提供一个新的靶点。
more >>
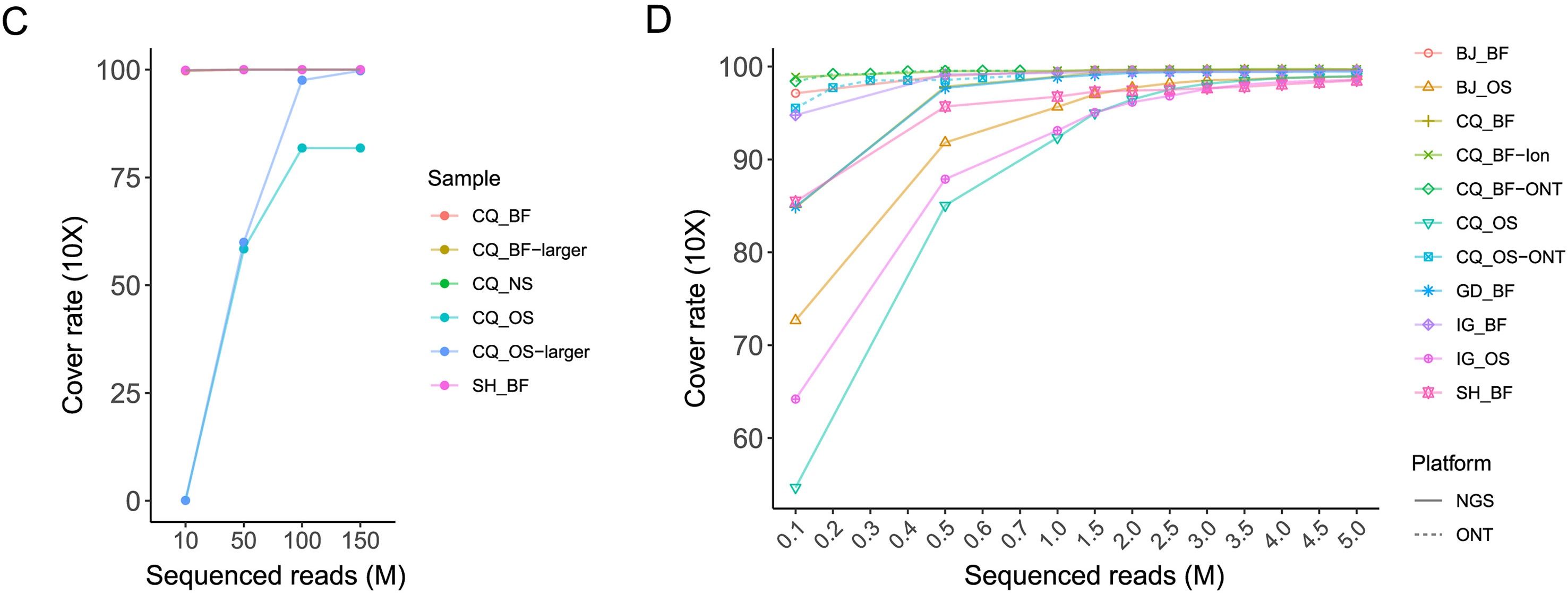
基于宏基因和扩增子测序快速测定临床样品中猴痘病毒完整基因组并进行分子溯源
猴痘病毒(MPXV)当前在全球范围内流行。基因组测序和分子溯源有助于诊断和控制猴痘病毒。猴痘病毒基因组较大、AT碱基偏倚严重且含有大量串联重复序列,从低病毒载量的临床样本中快速鉴定猴痘病毒完整基因组的策略需要不断地优化,以有效地追踪病毒遗传变异。针对来自中国五个猴痘病例的多个临床样本,本研究快速地测定了多个临床样本中猴痘病毒的完整基因组并准确组装了复杂串联重复序列。我们评估了mNGS、扩增子测序技术以及三种测序平台在猴痘病毒基因组测序方面的综合表现。扩增子测序可高效地对临床样本进行测序且成本较低,利于获得高质量的猴痘病毒基因组。三代测序策略可显著提升猴痘病毒基因组中末端串联重复区域的组装质量并纠正了已发表猴痘病毒基因组序列中一处常见的组装错误。此外,我们还在首个输入型猴痘病例感染的病毒中鉴定出了多个单核苷酸变异,提示猴痘病毒可在宿主体内快速演化。本研究成果对制定临床样品中猴痘病毒完整基因组的快速测定策略具有重要意义,将促进猴痘病毒变异及流行趋势监测工作。
more >>
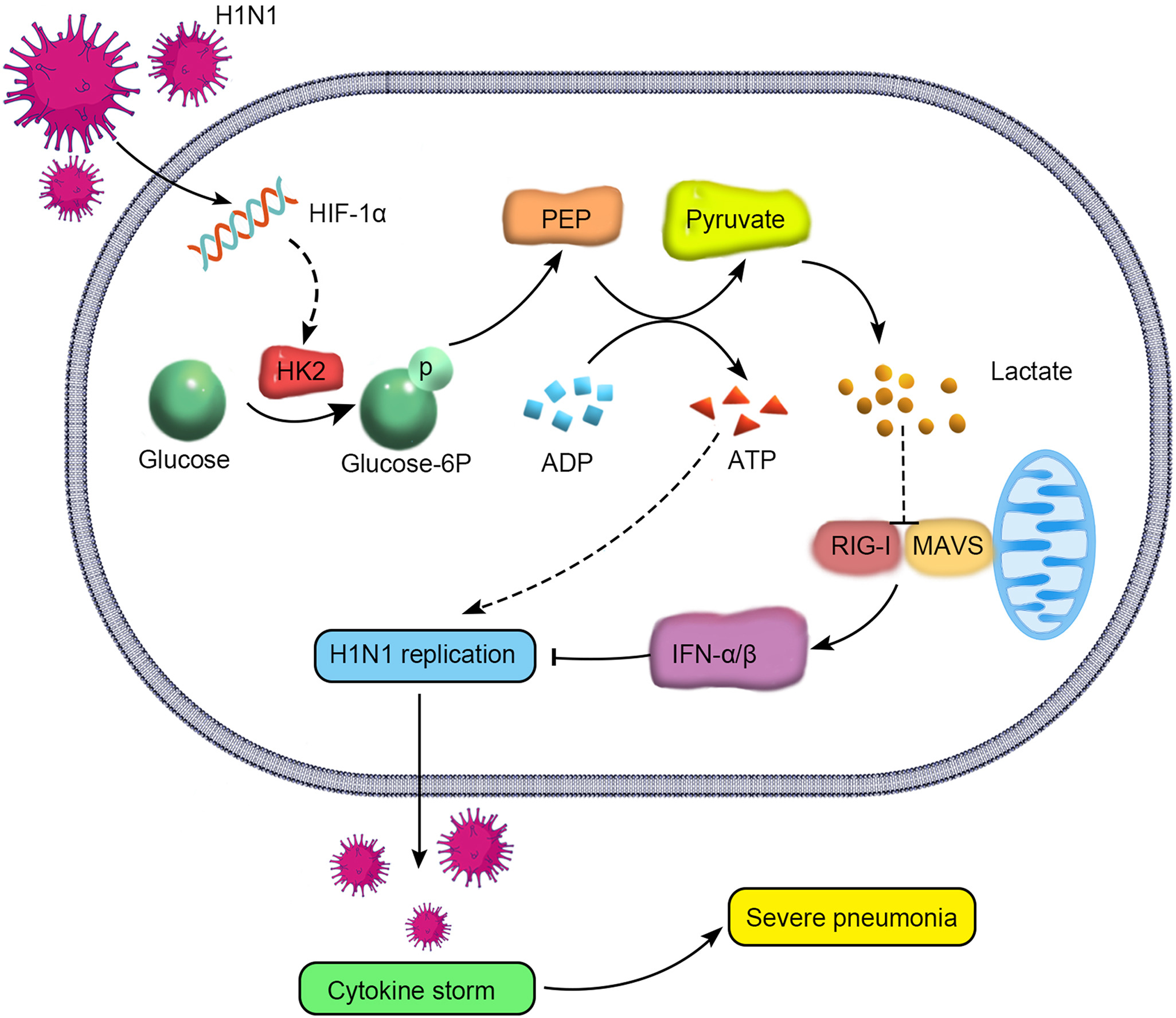
HIF-1α在H1N1病毒感染致重症肺炎中通过调控代谢重编程促进病毒复制及细胞因子风暴
H1N1病毒致重症肺炎患者的病死率与病毒复制及细胞因子风暴密切相关。然而,引起病毒复制和细胞因子风暴的机制仍未完全阐明。我们发现,HIF-1α活化参与调控H1N1病毒复制及细胞因子风暴。H1N1病毒感染后,HIF-1α的表达明显升高。体内外研究证实,抑制HIF-1α后,可以明显减轻H1N1病毒引起的肺损伤,减轻病毒复制及细胞因子风暴。其机制可能是,H1N1病毒感染肺泡上皮细胞后,可以诱导糖代谢向糖酵解偏移,快速产生ATP和乳酸;抑制糖酵解可以明显减少病毒复制及炎症因子。进一步研究发现,H1N1病毒能够通过活化HIF-1α促进糖酵解关键酶HK2的表达,一方面为病毒复制快速供能,另一方面产生的乳酸能够减少MAVS/RIG-I复合物的形成,进而抑制IFN-α/β的产生。综上所述,这些结果表明,H1N1感染后通过上调HIF-1α的表达调控HK2促进细胞代谢重编程向糖酵解偏移,增加病毒复制及细胞因子风暴。本研究将为寻找治疗H1N1病毒感染引起重症肺炎的潜在靶点提供理论依据。
more >>
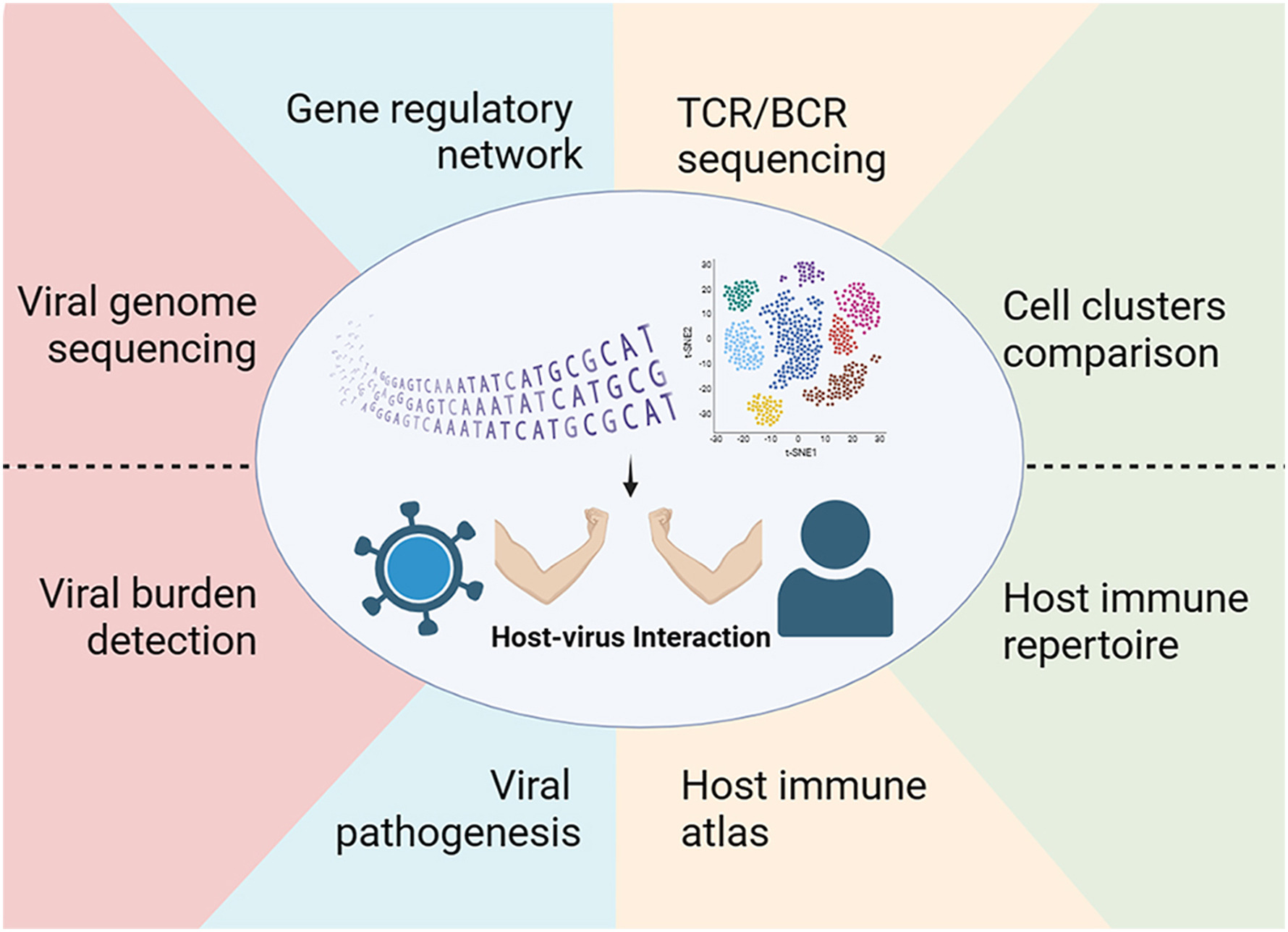
单细胞转录组测序技术揭示病毒-宿主相互作用
单细胞转录组测序(Single-cell RNA sequencing,scRNA-seq)是一种对分离的单个细胞进行转录组测序的新技术,目前已被用于分析单个细胞水平的病毒-宿主相互作用。本综述中,我们简要介绍了现有的scRNA-seq技术及其优缺点,并深入探讨了scRNA-seq在分析病毒发病机制、宿主免疫反应和疫苗评估方面的应用。最后,我们提出,未来scRNA-seq将广泛应用于病毒学研究。
more >>
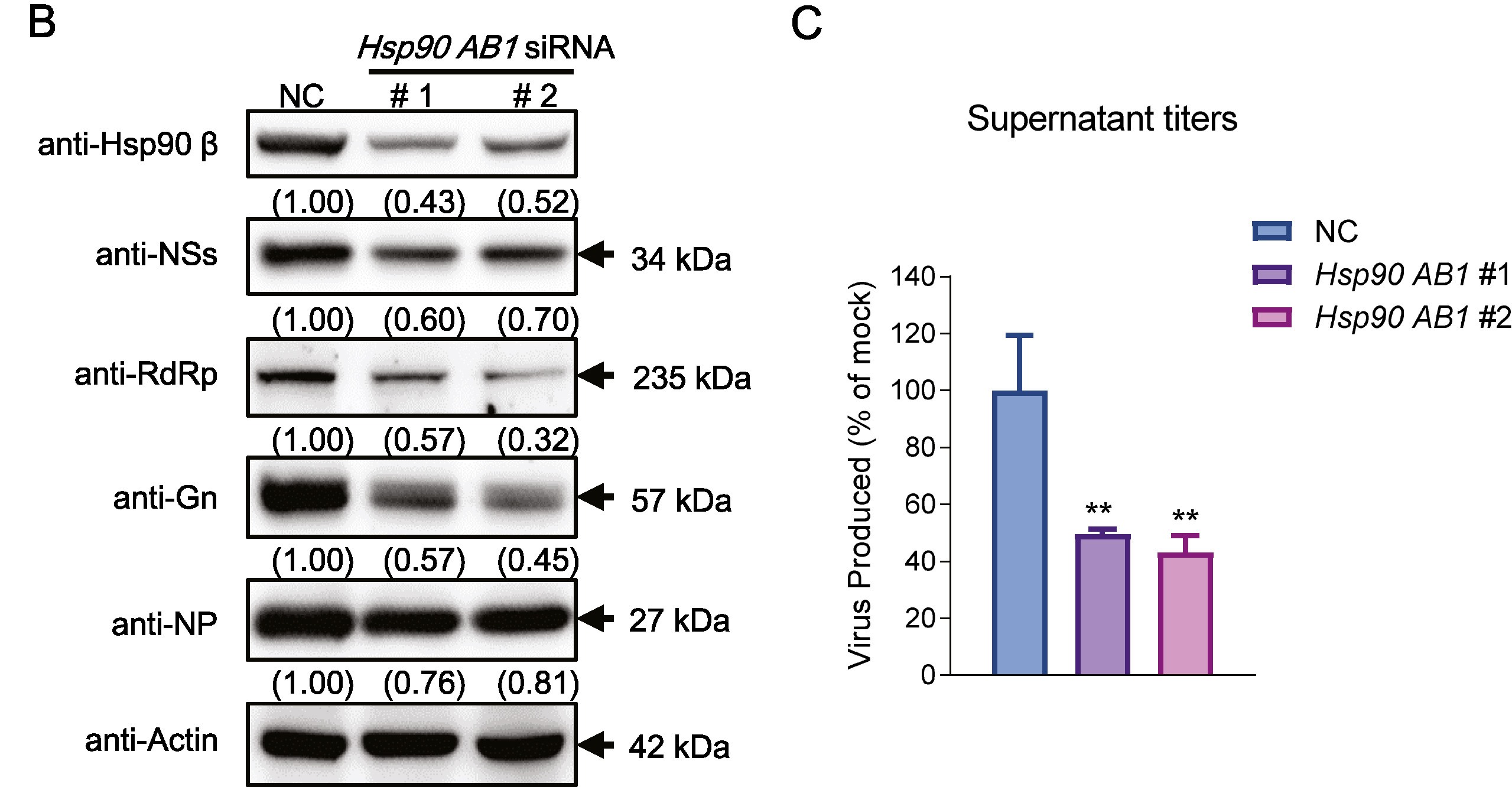
热休克蛋白90 β对发热伴血小板减少综合征病毒的感染至关重要
由发热伴血小板减少综合征病毒(SFTSV)感染引起的发热伴血小板减少综合征(SFTS)是流行于东亚地区的一种新发蜱传传染病,死亡率高达30%。目前关于SFTSV在感染/致病过程中与宿主的相互作用仍然知之甚少。热休克蛋白90(Hsp90)家族由多种高度保守的分子伴侣蛋白组成,在蛋白质的正确折叠和重塑中必不可少,因此对许多病毒的感染有广泛的影响。本研究表明,Hsp90是参与SFTSV感染的重要宿主因子。Hsp90抑制剂可显著降低SFTSV的复制、病毒蛋白表达和非结构蛋白(NSs)包涵体的形成。在4种病毒蛋白中,Hsp90抑制剂对NSs表达水平的降低最为显著,进一步我们通过测试病毒的转录情况,结果显示Hsp90抑制剂未影响基因组的转录,表明Hsp90抑制剂影响的是NSs蛋白翻译水平。进一步,NSs与Hsp90的四种异构体(Hsp90 α、Hsp90 β、GRP94及TRAP1)的免疫共沉淀实验显示只有Hsp90 β与NSs产生特异性相互作用,其他异构体不与其相互作用。同时,我们利用siRNA抑制Hsp90 β表达也能抑制SFTSV的复制。综上结果表明,Hsp90 β在SFTSV感染过程中起着关键作用,可能是开发抗SFTS药物的潜在靶点。
more >>
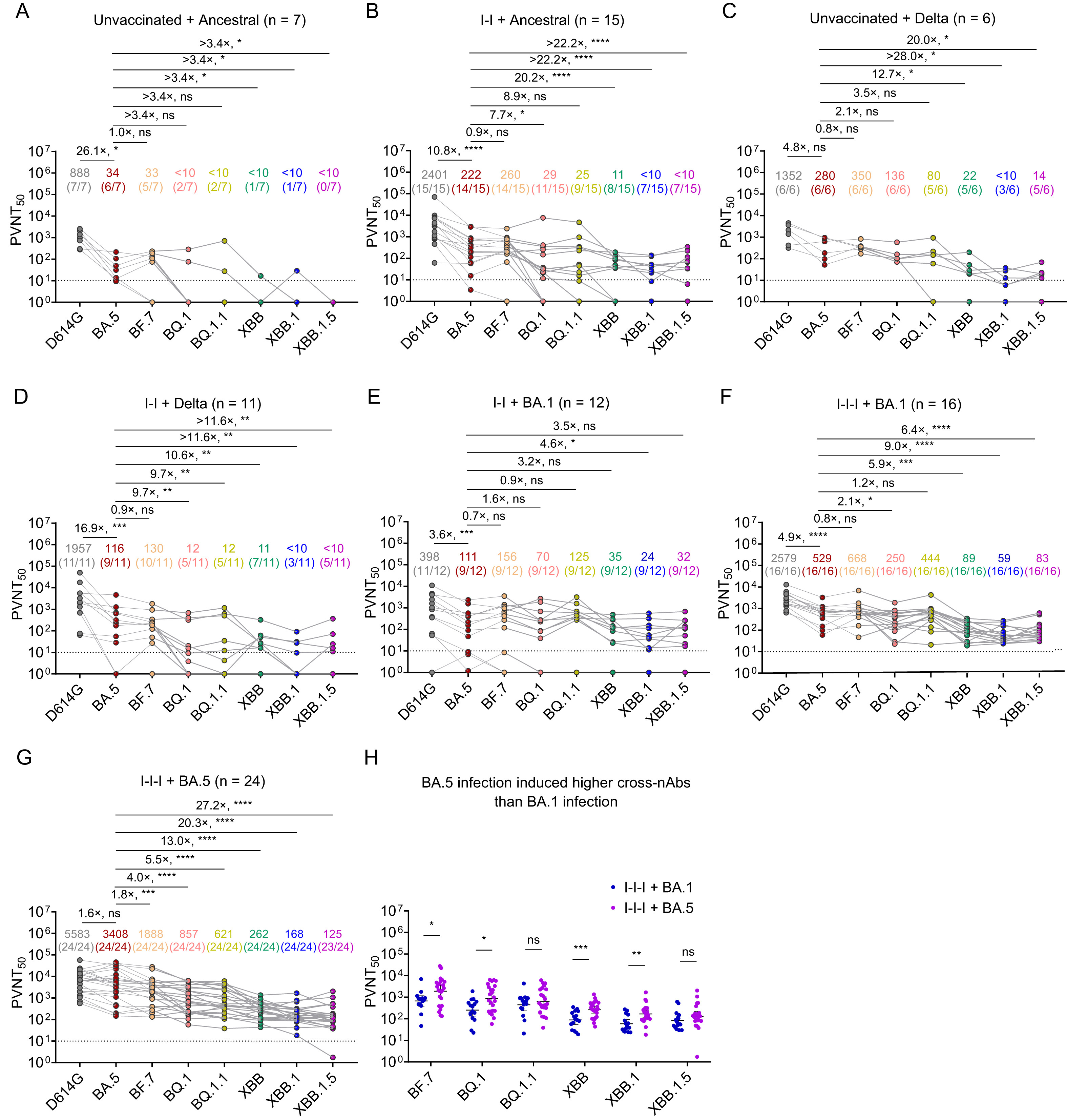
接种灭活疫苗后早期新冠病毒突破性感染者体内产生了极低滴度的针对当前奥密克戎XBB各种突变株的中和抗体
中国人群大多数都接种过新冠病毒灭活疫苗,但其中仍有相当一部分人在过去三年中还被新冠病毒感染。最近奥密克戎(Omicron) XBB突变株及其子代谱系已在我国流行,因此,急需了解此类人群对这些流行株的免疫保护水平。在本研究中,我们从云南省收集了91份曾经感染SARS-CoV-2原型株、Delta、Omicron BA.1或Omicron BA.5突变株的康复者血清样本,用基于VSV骨架的假病毒评估了它们针对当前流行的Omicron BF.7、BQ.1、BQ.1.1、XBB、XBB.1和XBB.1.5突变株的的中和活性。结果发现:(1)在未接种过疫苗和接种两剂灭活疫苗后,再被新冠病毒原始株、Delta株或BA.1株感染的人群中,其恢复期血清几乎检测不到针对当前这些Omicron流行株的中和抗体;(2)在接种三剂灭活疫苗后,再被BA.1或BA.5株感染的人群中,其恢复期血清针对当前Omicron变异株的中和抗体滴度升高;并且,相比BA.1株,BA.5株感染诱导了较高滴度的中和抗体;(3)在检测的所有恢复期血清中,都发现针对XBB、XBB.1和XBB.1.5的中和抗体比针对BF.7、BQ.1和BQ.1.1的中和抗体滴度低。本研究表明:在接种新冠病毒灭活疫苗后,再被早期流行的新冠病毒株感染的人群体内,尚未产生足够的交叉中和抗体,以抵御当前流行的Omicron新型突变株的再次感染。因此,此类人群需要加强个人防护;同时,也提示进一步研发新型广谱疫苗的必要性。
more >>
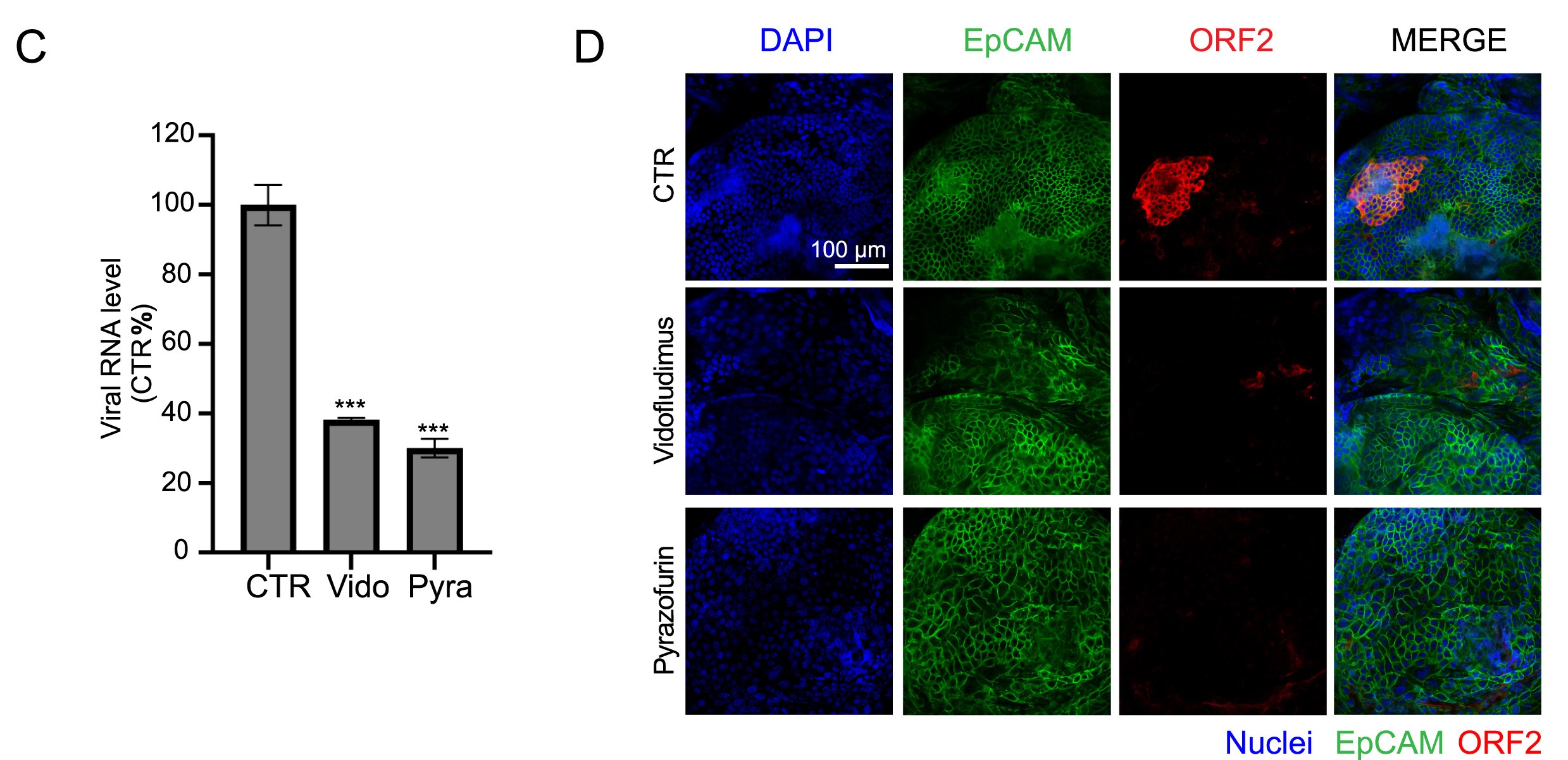
药物筛选发现维多氟拉迪莫和吡唑霉素是治疗戊型肝炎的新型候选药物
戊型肝炎病毒(hepatitis E virus,HEV)感染可引起严重的并发症和高死亡率,尤其是在孕妇、器官移植受者、或肝病患者和免疫抑制患者中。然而,目前仍缺乏治疗慢性戊型肝炎病毒感染的特定疗法。本研究通过筛选262种药物/化合库鉴定发现vidofludimus calcium和pyrazofurin为新型抗HEV小分子药物。Vidofludimus calcium是新一代二氢乳清酸脱氢酶(DHODH)抑制剂,用于治疗自身免疫性疾病或SARS-CoV-2感染(临床二期、三期)。Pyrazofurin选择性靶向尿苷单磷酸合成酶(UMPS)。在一系列细胞培养模型和不同个体人原代肝脏类器官模型中vidofludimus calcium和pyrazofurin都具有显著的抗HEV作用。并且两种药物均能高效抑制野生型HEV菌株和利巴韦林治疗失败相关的HEV毒株(Y1320H,G1634R)。令人鼓舞的是,针对HEV这两种药物都表现出相当大的的治疗窗口。例如,vidofludimus calcium的抑制HEV复制的IC50值比目前临床应用给患者的治疗剂量低4.6--7.6倍。两种药物的抗HEV作用机制是通过阻断宿主细胞嘧啶的从头合成通路。此外,vidofludimus calcium和pyrazofurin与IFN-α联合用药可产生协同抗病毒功效。总之,本研究确定vidofludimus calcium和pyrazofurin是治疗HEV感染的有效候选药物。基于其抗病毒效力,以及临床研究中确定的良好安全性,本研究结果支持启动临床研究,将这些药物用于治疗慢性戊型肝炎。
more >>
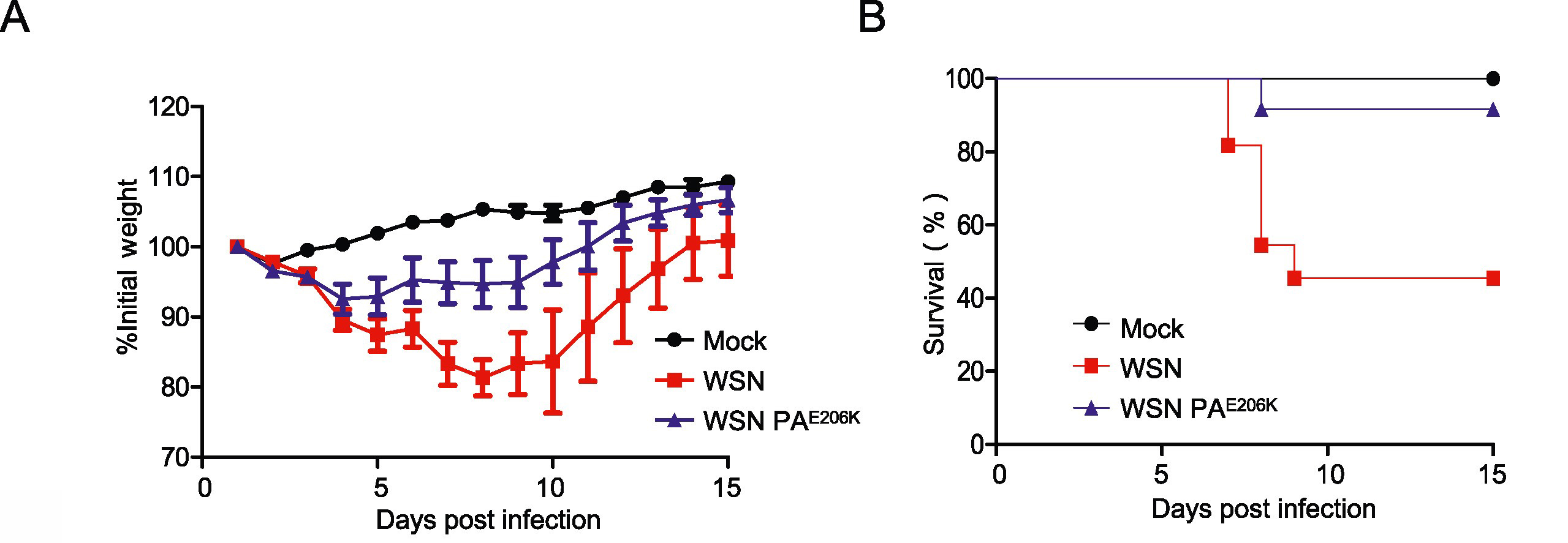
H1N1流感病毒中自然发生的PA E206K点突变在高温下不利于流感病毒的复制
2009年4月,甲型H1N1流感病毒的出现标志着21世纪疾病的第一次大流行。在这项研究中,我们观察到从中国和日本患者分离到的两个2009甲型H1N1流感病毒株的聚合酶活性存在明显的差异。病毒RNA依赖性RNA聚合酶复合物的三个主要蛋白质亚基(PB2、PB1和PA)的序列比对和随后的突变分析表明,PA蛋白的单个氨基酸的突变(E206K)是影响流感病毒复制发生改变的原因。进一步的实验表明,PAE206K的存在降低了A/WSN/33在哺乳动物细胞中的复制能力,同时也降低了流感病毒在体内的致病性。而分子机制研究表明,PA E206K是一种温度敏感的突变,在高温(39.5℃)下无法将PB1-PA复合物转运到细胞核。因此,PA蛋白中这种天然存在的变体是开发减毒流感活疫苗的候选突变。
more >>
H9N2的HA和PA蛋白中的小鼠适应性突变对病毒复制起相互拮抗作用
A型流感病毒(influenza A)中,禽源H9N2病毒具有广泛的宿主范围。然而,我们对H9N2病毒在哺乳动物中的适应基础的了解有限。为了揭示禽源H9N2适应哺乳动物的分子基础,我们在小鼠中对一株禽源A/Chicken/Hunan/8.27 YYGK3W3-OC/2018(H9N2)(3W3)毒株进行了肺部连续传代,在HA和PA蛋白中共鉴定到6个氨基酸突变位点。体内实验结果表明,HA-L226Q、T511I和A528V(H3 numbering)突变可增强病毒对小鼠的致病性和增加病毒在小鼠肺部的复制,其中,HA-L226Q是关键决定因素。PA突变(PA-T97I、I545V及S594G)可增强病毒在哺乳动物细胞中的聚合酶活性、增加病毒在体外和体内的复制水平。PA-T97I通过促进病毒聚合酶复合物组装以促进病毒聚合酶活性。我们的研究结果显示,PA-97I和/或PA-545V与3个HA点突变拮抗并影响病毒复制。此外,当2个和3个PA点突变与HA-226Q结合时,表现出对病毒复制的拮抗作用。值得注意的是,PA突变与双点HA突变的任何组合都会拮抗病毒复制。我们还观察到PA-545V和PA-97I组合、以及HA-528V和PA-545V组合分别对病毒复制有拮抗作用。我们的结果表明,HA和PA蛋白中的几种拮抗突变组合可影响病毒复制。这些相互拮抗的突变可能有助于H9N2病毒适应小鼠和哺乳动物细胞。这些发现对理解禽源H9N2适应哺乳动物分子基础和评估其在哺乳动物中的危害提供了新的数据支撑。
more >>
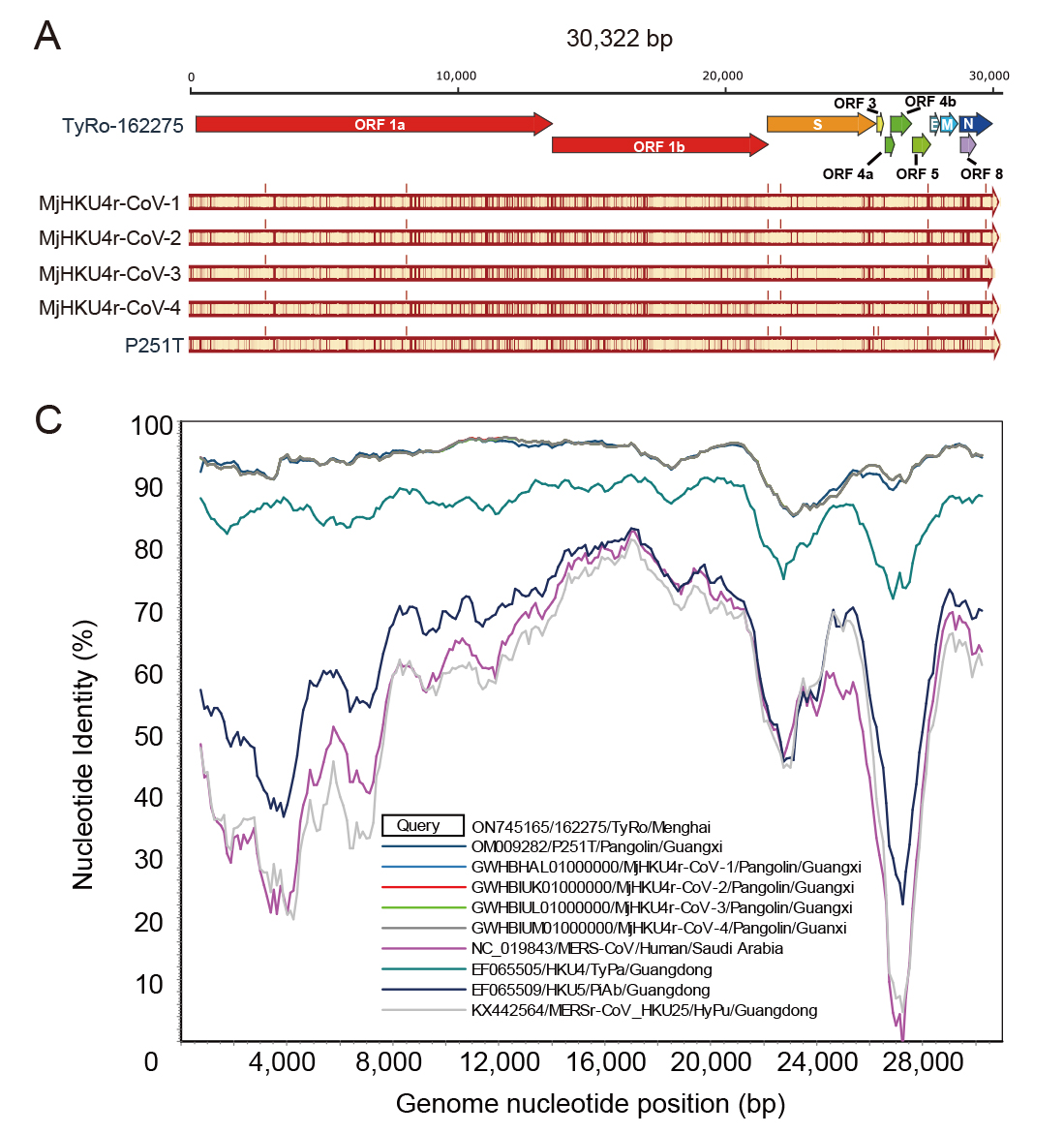
中国南方褐扁颅蝠中发现穿山甲冠状病毒HKU4r-CoV
野生动物冠状病毒(CoV)外溢事件目前受到公众的广泛关注,这加剧了对携带有多种病毒的蝙蝠开展冠状病毒调查的必要性。本研究在2016年和2017年期间共采集了位于中国云南省和广东省的9个地点20种蝙蝠的729份肛拭子样品,分析了样品中α冠状病毒和β冠状病毒的分子特征和遗传多样性。通过RT-PCR检测,在分布于6个地点的9种蝙蝠中发现58种(8.0%)冠状病毒。进一步利用Illumina测序平台获得了其中来自褐扁颅蝠(Tylonycteris robustula)的2个冠状病毒TyRo-CoV-162275和TyRo-CoV-162269的全长基因组序列。对这2个基因组的序列分析表明,TyRo-CoV-162275与马来亚穿山甲(Manis javanica)的冠状病毒MjHKU4r-CoV一致性最高(93.9%);而TyRo-CoV-162269与贵州的褐扁颅蝠的HKU33-CoV一致性最高(94.1%)。更重要的是,在TyRo-CoV-162275的S蛋白序列中发现了furin蛋白酶切割位点,揭示该病毒可能与MERS-CoV一样,以二肽基肽酶-4(hDPP4)为受体感染人体细胞。这是首次对含furin蛋白酶切割位点的蝙蝠冠状病毒HKU4r-CoV的报道。这些发现拓展了我们对冠状病毒地理和宿主分布的认知。
more >>
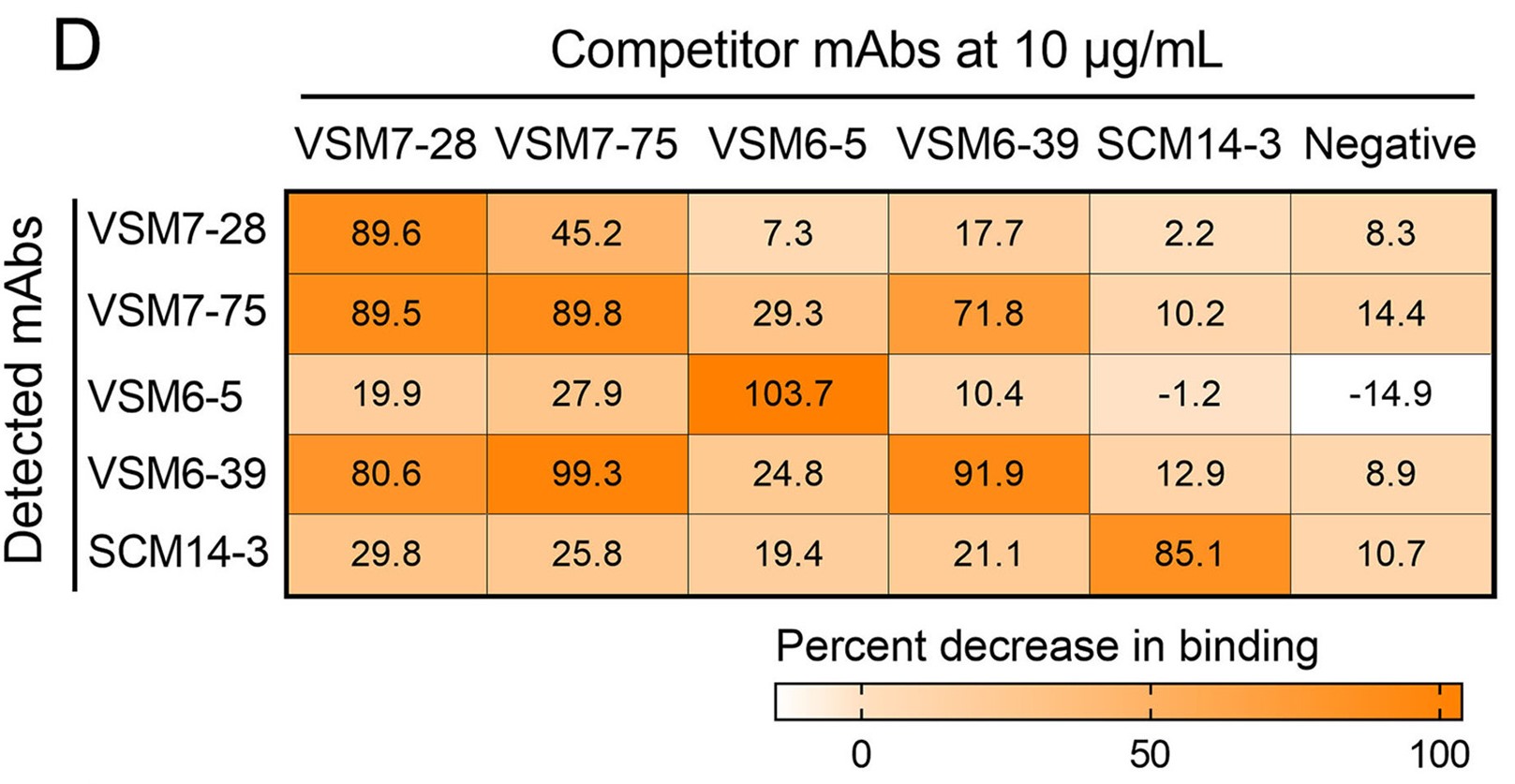
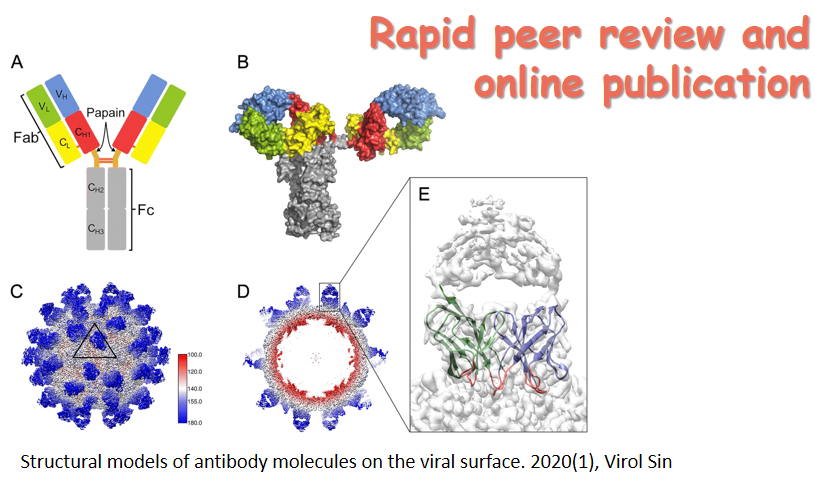
泛冠状病毒棘突蛋白S2特异性单克隆抗体的分离与鉴定
在不到20年的时间里,动物冠状病毒向人类的溢出引起了三次严重公共卫生事件:严重急性呼吸综合征(SARS)、中东呼吸综合征(MERS)和新型冠状病毒肺炎(COVID-19)。冠状病毒主要通过表面棘突蛋白与宿主受体蛋白相互作用而感染宿主细胞。此前,我们从COVID-19康复者和接种灭活疫苗的人群中获取了274个针对新型冠状病毒棘突蛋白的单克隆抗体。在此,我们对这些抗体进行深入的分析,旨在筛选识别泛冠状病毒的单克隆抗体。结果发现了5个对25种来自人和动物的冠状病毒具有广谱反应性的单克隆抗体,包括7种人冠状病毒、新型冠状病毒的突变株,来自果子狸、蝙蝠和穿山甲的异常冠状病毒,以及牛、山羊和鸡的冠状病毒。进一步对这些抗体的抗病毒特征进行分析,发现抗体VSM7-28、VSM7-75和VSM6-39靶向冠状病毒棘突蛋白融合肽1表位,另外两个单克隆抗体靶向不同但保守的表位。5个抗体的重链和轻链都经历了高水平的体细胞突变,这对它们的广谱结合活性至关重要。这些单克隆抗体的发现为未来开发未知冠状病毒感染监测试剂和广谱疫苗奠定了基础。
more >>
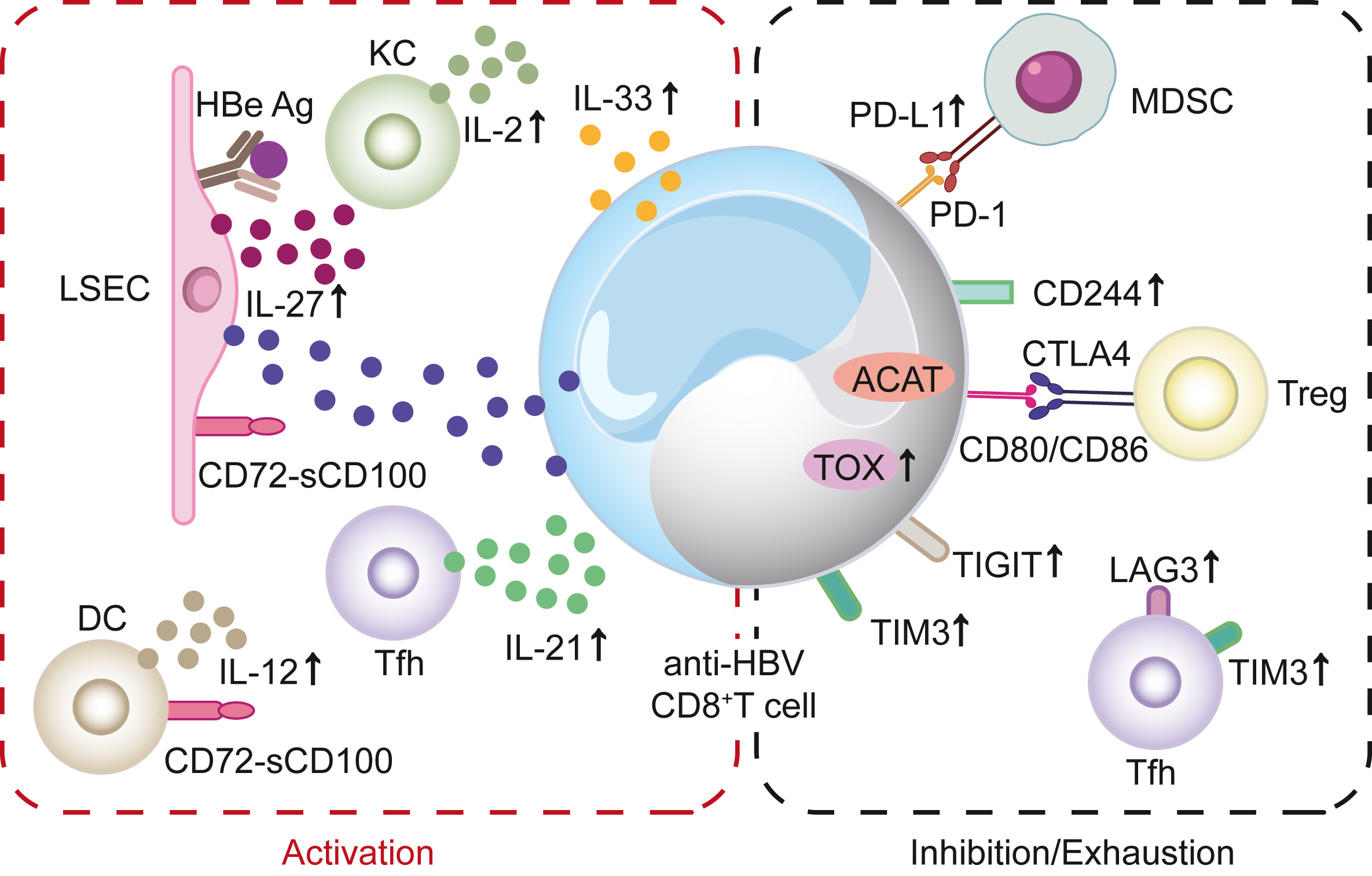
HBV感染状态T细胞活化及耗竭机制的研究进展
慢性乙型肝炎病毒(HBV)感染仍然是全球主要的公共卫生问题,T细胞应答在介导HBV清除方面起着关键作用。因此,关于HBV特异性T细胞从活化到耗竭的特征研究进展迅速。我们通过回顾过去五年发表的基础和临床研究,总结了T细胞免疫在HBV感染状态的最新进展。我们全面总结了有效诱导抗HBV特异性T细胞免疫应答的机制,和慢性HBV感染中T细胞功能障碍的最新进展。此外,我们简要介绍了当前关于恢复抗HBV特异性T细胞免疫应答的新治疗策略。
more >>
新冠病毒反向遗传系统:发展与应用
严重急性呼吸综合征冠状病毒2(新冠病毒;SARS-CoV-2)引起的新冠大流行对人类健康造成了严重危害,也影响全球经济。反向遗传系统为SARS-CoV-2的科学研究提供了非常有用的帮助。研究人员通过人工操纵病毒基因组序列,可以获得经修饰的全长感染性克隆、表达报告基因的重组病毒以及能够在生物安全2级实验室操作的非感染性病毒复制子。这些工具在研究病毒的分子生物学特性、筛选抗病毒药物以及促进候选减毒活疫苗的研发方面发挥了重要作用。本文详细总结了SARS-CoV-2反向遗传系统的构建策略、发展历程和具体应用,以期能为其它冠状病毒的研究提供一定的参考。
more >>
优先发表栏目展示本刊经同行评议确定正式录用的文章,这些文章目前处在编校过程,尚未确定卷期及页码,但可以根据DOI进行引用。
显示方式:

2024, 39(2): 177-193.
doi: 10.1016/j.virs.2024.01.007
收稿日期: 2023-06-20
录用日期: 2024-01-18
出版日期: 2024-01-23

由猴痘病毒引起的持续流行的人类猴痘引起公众对猴痘病毒及其他痘病毒的未来传播的关注。猴痘病毒是一种典型的人兽共患病毒,可以感染人类并引起类似天花的症状。猴痘病毒属于痘病毒科,痘病毒科宿主范围较广,从节肢动物到脊椎动物均可感染。不同宿主间痘病毒的跨物种传播经常被报道并引发流行病。痘病毒具有复杂的线性双链DNA基因组,编码数百种蛋白质。与痘病毒宿主范围相关的基因称为宿主范围基因。本综述简要介绍了痘病毒的分类、系统发生和宿主,并全面总结了目前痘病毒跨物种传播的知识。痘病毒的宿主范围基因尤其被重点描述并深入讨论了它们对病毒宿主范围的影响。我们期望这篇综述对痘病毒的跨物种传播以及宿主范围基因目前的研究进展提供一个全面的描述,为学术研究和未来疾病控制提供宝贵的参考。

2024, 39(2): 194-204.
doi: 10.1016/j.virs.2024.02.003
收稿日期: 2023-10-09
录用日期: 2024-02-08
出版日期: 2024-02-13
在东亚常见的长角血蜱可以传播多种致病病毒,包括严重急性发热伴血小板减少综合症病毒(SFTSV)。本研究调查了2019年至2020年间在湖北省三个山区内吸血和未吸血的长角血蜱的病毒组。测序分析鉴定出与参考病毒相关的39个病毒序列。这些病毒序列属于未分类病毒和七个病毒家族:Chuviridae、Nairoviridae、Orthomyxoviridae、Parvoviridae、Phenuiviridae、Rhabdoviridae和Totiviridae。通过生物信息学分析,我们探讨了影响蜱虫携带的病毒组成结构的关键因素。此外,基于全基因组序列的系统发育分析阐明了Henan tick virus(HNTV)、Dabieshan tick virus(DBSTV)、Okutama tick virus(OKTV)和Jingmen tick virus(JMTV)的分子进化特征。基于单只蜱的精细分子流行病学调查表明DBSTV是长角血蜱中最常见的病毒,流行率为12.59%,其次是HNTV(0.35%),但未检测到JMTV和OKTV。这些结果加深了对中国中部地区长角血蜱病毒组的了解,提示蜱虫吸血状态和地理位置对蜱病毒组结构组成方面的影响作用。对本研究中新发现的病毒株,将在后续的调查和研究中加强对这些病毒的监测,全面评估其溢出潜力和对公共卫生的潜在影响。

2024, 39(2): 205-217.
doi: 10.1016/j.virs.2024.02.002
收稿日期: 2023-10-03
录用日期: 2024-02-06
出版日期: 2024-02-10
猪被认为是流感病毒的“中间宿主”或“混合容器”,可以产生具有大流行潜力的毒株。从2020年到2021年,我们在中国南部的广东、云南和贵州以及中国北部的河南和山东的养猪场进行了猪H1N2流感(swH1N2)病毒监测。我们系统地分析了swH1N2病毒的进化和致病性,并对其复制和传播能力进行了表征。分离的病毒是四重重组H1N2病毒,含有来自pdm/09 H1N1 (PB2、PB1、PA和NP基因)、三重重组猪流感(NS基因)、欧亚禽类(HA和M基因)和最近人源H3N2 (NA基因)谱系的基因。SW/188/20和SW/198/20的NA、PB2和NP与A/Guangdong/Yue Fang277/2017 (H3N2)具有较高的基因相似性。swH1N2的HA基因具有较高的进化速率。分离的5株swH1N2病毒在人、犬和猪细胞以及小鼠的鼻甲、气管和肺中都能有效地复制。A/swine/Shandong/198/2020在猪的呼吸道中可有效复制,并在猪群中有效传播。总的来说,这些当前流行的swH1N2病毒具有人畜共患的潜力,强调了加强swH1N2病毒监测的必要性。

2024, 39(2): 218-227.
doi: 10.1016/j.virs.2024.01.010
收稿日期: 2023-10-18
录用日期: 2024-01-31
出版日期: 2024-02-03
严重急性呼吸系统综合症冠状病毒2型(SARS-CoV-2)欧密克戎(Omicron)变异株因其高传播性而臭名昭著,但对其亚基因组RNA(sgRNA)的表达知之甚少。本研究应用RNA高通量测序技术描绘了118例欧密克戎BA.2变异株(Omicron BA.2)和338例非关注D614G变异株(non-VOC-D614G)的典型sgRNA的定量和定性特征。结果显示,无论患者的性别、年龄以及是否患有肺炎,Omicron BA.2和non-VOC-D614G均表现出由9个典型sgRNA相对丰度组成的独特特征定量表达谱。这种表达谱在病毒载量低时会消失,表明sgRNA模式有可能用于指示特定时间点患者的病毒活性。对Omicron BA.2和non-VOC-D614G典型sgRNA的特征定性表达谱的描述发现,所有9个典型sgRNA均表达的模式与病毒载量具一致相关性(AUC = 0.91,95% CI 0.88 - 0.94),其中的sgRNA ORF7b可被视为最佳替代标志物,用于检测患者个体化感染状况。sgRNA在疫苗和抗病毒药物开发中的应用潜力值得进一步挖掘。

2024, 39(2): 228-234.
doi: 10.1016/j.virs.2024.03.002
收稿日期: 2023-08-26
录用日期: 2024-03-05
出版日期: 2024-03-08
瓜伊库蚊病毒(GCXV)是一种新分离鉴定的从中美洲和南美洲库蚊分离获得的分节段病毒,其基因组由4到5个单股正链RNA片段组成。目前,GCXV在蚊子体内的感染动力学和传播能力尚不清楚。在本研究中,我们首先使用反向遗传学方法在C6/36细胞中拯救了分别包含4个和5个RNA片段的两种GCXVs(4S和5S)。体外生物学鉴定进一步表明,两种GCXV表现出相当的复制动力学、蛋白表达能力和病毒滴度。重要的是,将两种GCXV经口感染致倦库蚊,在感染后4-10天的身体、唾液腺、中肠和卵巢中均检测到GCXV特异的RNA。此外,两种GCXV可定植于蚊卵,导致第二个生殖腺营养周期的阳性率为15%-35%。综上所述,我们的研究结果表明,在经口感染后的第一和第二性腺营养周期的致倦库蚊卵中,均可检测到4或5个RNA片段的GCXVs。

2024, 39(2): 235-250.
doi: 10.1016/j.virs.2023.12.001
收稿日期: 2023-11-18
录用日期: 2023-12-03
出版日期: 2023-12-09
呼吸道合胞病毒(Respiratory syncytial virus, RSV)是引起全世界婴幼儿急性下呼吸道感染(acute lower respiratory tract infection, ALRTI)最重要的病原体。RSV的包涵体(inclusion bodies, IBs)通过液-液相分离(liquid-liquid phase separation, LLPS)形成, 其内部结构——包涵体相关颗粒(IB-associated granules, IBAGs),瞬时浓缩新合成的病毒mRNAs及转录抗终止因子M2-1,但是IBAGs的形成机制以及如何调控病毒mRNAs翻译的分子机制仍不明确。本研究发现RSV IBs的内部结构实际上是由次级LLPS形成的、不含M2-1的病毒信使核糖核蛋白(messenger ribonucleoprotein, mRNP)凝聚物。机制上,RSV核蛋白(nucleoprotein, N)和M2-1与PABP相互作用将PABP募集至RSV IBs,促使PABP通过RNA识别基序(RNA-recognition motif, RRM)结合IBs内转录的病毒mRNAs并驱动次级相分离,即PABP是驱动次级相分离的支架蛋白。此外,PABP-eIF4G1相互作用调控病毒mRNP凝聚物的组成,将特异的翻译起始因子(eIF4G1、eIF4E、eIF4A、eIF4B 呼吸道合胞病毒(Respiratory syncytial virus, RSV)是引起全世界婴幼儿急性下呼吸道感染(acute lower respiratory tract infection, ALRTI)最重要的病原体。RSV的包涵体(inclusion bodies, IBs)通过液-液相分离(liquid-liquid phase separation, LLPS)形成, 其内部结构——包涵体相关颗粒(IB-associated granules, IBAGs),瞬时浓缩新合成的病毒mRNAs及转录抗终止因子M2-1,但是IBAGs的形成机制以及如何调控病毒mRNAs翻译的分子机制仍不明确。本研究发现RSV IBs的内部结构实际上是由次级LLPS形成的、不含M2-1的病毒信使核糖核蛋白(messenger ribonucleoprotein, mRNP)凝聚物。机制上,RSV核蛋白(nucleoprotein, N)和M2-1与PABP相互作用将PABP募集至RSV IBs,促使PABP通过RNA识别基序(RNA-recognition motif, RRM)结合IBs内转录的病毒mRNAs并驱动次级相分离,即PABP是驱动次级相分离的支架蛋白。此外,PABP-eIF4G1相互作用调控病毒mRNP凝聚物的组成,将特异的翻译起始因子(eIF4G1、eIF4E、eIF4A、eIF4B和eIF4H)募集至次级凝聚相中。最后,本研究发现病毒mRNP凝聚物的功能是对病毒mRNAs进行活化,促进病毒mRNAs对核糖体的募集,从而提高病毒mRNAs的翻译效率。我们的研究揭示了一种新的、由LLPS调控的病毒蛋白翻译机制,提供了一种新的靶向次级凝聚相的抗病毒策略。

2024, 39(2): 251-263.
doi: 10.1016/j.virs.2024.01.002
收稿日期: 2023-08-09
录用日期: 2024-01-09
出版日期: 2024-01-14
病毒性脑炎仍然是全球公共卫生安全的重大威胁。在课题组前期研究中,我们发现拉斯穆森脑炎(Rasmussen’s encephalitis,RE)患者的脑组织中IFN-β、STING和IFI16等多种抗病毒关键基因的表达量显著降低。这种脑炎是一种罕见的慢性神经系统疾病,好发于儿童,其典型临床特征是单侧半球脑萎缩。此外,课题组还发现人类疱疹病毒(Human herpes viruses,HHVs)的累积病毒感染评分与RE的单半球萎缩显著正相关。I型干扰素(IFN-I)信号通过与IFN-α/β受体(IFNAR)结合发挥作用,对抗感染固有免疫至关重要。在本研究中,我们通过单侧眼周注射使野生型(Wild type,WT)小鼠及IFNAR缺陷的A6小鼠感染单纯疱疹病毒1型(Herpes simplex virus 1,HSV-1),以探究IFN-I信号传导与HHVs诱导的脑损伤之间的关系。虽然所有小鼠脑组织都表现出典型的病毒性脑炎病变,但仅有A6小鼠表现出HSV诱导的癫痫。我们进一步对小鼠脑组织进行RNA-Seq分析,并通过功能富集分析和蛋白质-蛋白质相互作用网络揭示了四种与HSV诱导的癫痫症状呈正相关的基因模块。此外,我们还鉴定了10与癫痫发作关联最为密切的关键基因。本研究表明,IFN-I信号通路可以有效抑制HHVs诱导的神经症状和脑组织病理损害,从而证实了RE和其他HHVs脑炎中IFN-I信号通路激活不足与脑萎缩之间的正相关性。

2024, 39(2): 264-276.
doi: 10.1016/j.virs.2024.01.005
收稿日期: 2023-07-01
录用日期: 2024-01-15
出版日期: 2024-01-23
猪繁殖与呼吸综合征病毒(PRRSV)是一种对经济产生严重破坏的主要病原体,已经进化出多种策略以逃避先天免疫。通过胞质内黑色素瘤分化相关基因5(MDA5)-一种感知病毒RNA的受体来降低抗病毒干扰素的表达,从而在很大程度上助长了PRRSV的免疫逃逸。在本研究中,我们观察到猪MDA5在PRRSV感染中转录和表达水平下调,但具体机制仍然不清楚。PRRSV感染细胞中,受到上调的激酶CK2α的作用,强化了自噬受体P62的磷酸化修饰并促进其与MDA5的相互作用,且E3泛素连接酶TRIM21引起的猪MDA5的K63泛素化,从而触发了经典的P62介导的自噬。此外,猪MDA5与含TCP1亚基2(CCT2)的分子伴侣相互作用,PRRSV编码的 nsp3促进它们的结合,促进了MDA5-CCT2-nsp3聚集体的形成和自噬清除,这一过程不依赖泛素。总之,PRRSV感染中通过两种自噬途径增强了MDA5的降解,包括MDA5与自噬受体P62以及聚集体自噬受体CCT2的结合,触发了强烈的先天免疫抑制。本研究揭示了PRRSV感染中免疫逃避的一种新机制,并为开发新疫苗或治疗药物提供了基础见解。

2024, 39(2): 277-289.
doi: 10.1016/j.virs.2024.01.003
收稿日期: 2023-10-17
录用日期: 2024-01-16
出版日期: 2024-01-20
甲型流感病毒(Influenza A virus, IAV)通过结合细胞表面唾液酸(Sialic acid,SA)受体进入宿主细胞,这是启动感染、传播和致病的关键步骤。了解导致IAV高效进入人类细胞的因素将有助于阐明病毒进入和致病的机制,并提供新的干预靶点。在本研究中,我们报道了一种新的膜蛋白C1QTNF5,它与IAV的血凝素(Hemagglutinin,HA)蛋白结合并在体内外促进IAV感染。鉴定出IAV血凝素的HA1区域是与C1QTNF5蛋白相互作用的关键区域,而C1QTNF5主要通过其N端(1- 103aa)与血凝素相互作用。此外,我们进一步发现过表达C1QTNF5可促进IAV进入,而阻断C1QTNF5和IAV血凝素之间的相互作用极大地抑制了病毒的进入。然而,在唾液酸缺陷的CHO-Lec2细胞中,C1QTNF5不能作为独立受体介导IAV感染,而是促进IAV附着在这些细胞上,这表明C1QTNF5是IAV的重要吸附因子。这项工作揭示了C1QTNF5作为一种新的IAV吸附因子,为抗病毒策略提供了新的视角。

2024, 39(2): 290-300.
doi: 10.1016/j.virs.2024.02.001
收稿日期: 2023-05-05
录用日期: 2024-01-31
出版日期: 2024-02-06
柯萨奇病毒B3(CVB3)是引起手足口病(HFMD)的病原体,其临床表现从轻到重不等。然而传统的CVB3感染小鼠研究主要导致病毒性心肌炎和胰腺炎,无法复制HFMD症状。这限制了我们对于CVB3病毒-宿主相互作用的理解。尽管叙利亚仓鼠和恒河猴已被广泛用于评估许多肠道病毒,但尚未有关于CVB3的系统报道。在本研究中,我们首次测试了叙利亚仓鼠通过不同途径对CVB3感染的敏感性。我们的研究发现,无论是腹腔注射还是鼻滴,都有效地使CVB3感染叙利亚仓鼠,导致典型的HFMD症状、鼻咽部定植和急性严重病理性损伤。值得注意的是,鼻滴组导致了更长的排毒周期和更严重的病理性损伤。在后续研究中,通过鼻滴感染CVB3的恒河猴也表现出HFMD症状、排毒、血清抗体转换、病毒核酸和抗原以及特定器官的病理性损伤,如心脏。令人惊讶的是,心肌酶水平没有显著差异。并且临床表现与常见轻症感染相似。总之,本研究建立了CVB3重症叙利亚仓鼠和轻症恒河猴HFMD模型,为理解病原学、预试验预防与评估和暴露后干预奠定了基础。

2024, 39(2): 301-308.
doi: 10.1016/j.virs.2024.02.006
收稿日期: 2023-07-05
录用日期: 2024-02-26
出版日期: 2024-03-05
手足口病(HFMD)是一种常见的儿科疾病,主要由肠道病毒引起,肠道病毒是重要的人类病原体。目前,还没有可用的抗病毒药物治疗肠道病毒感染。本研究利用EV-A71-eGFP报告病毒开发了一种优良的高内涵抗病毒筛选系统。基于该系统,我们筛选了包含1042种天然化合物的药物库,以鉴定潜在的EV-A71抑制剂。防己诺林碱(FAN)是一种双苄基异喹啉类生物碱,对引起手足口病的EV-A71、CV-A10、CV-B3和CV-A16等多种肠道病毒具有抑制作用。进一步的研究表明,FAN靶向肠道病毒生命周期的早期阶段。通过进一步筛选抗FAN的EV-A71耐药病毒株,我们证明VP1的两个突变(E145G和V258I)可使病毒对FAN产生抗性,推测VP1蛋白可能是FAN的潜在靶点。我们的研究表明,FAN是一种有效的EV-A71抑制剂,具有进一步成为一种广谱抗肠道病毒药物的潜力。

2024, 39(2): 309-318.
doi: 10.1016/j.virs.2024.03.001
收稿日期: 2023-08-11
录用日期: 2024-02-27
出版日期: 2024-03-06
新冠病毒(SARS-CoV-2)感染诱导的过度炎症是新冠肺炎的关键致病因素。我们和其他人的研究表明,肥大细胞在新冠病毒诱发过度炎症中起着至关重要的作用。我们之前观察到新冠病毒感染会导致人源化小鼠支气管周围和支气管-肺泡管交界处肥大细胞的积聚;此外,发现刺突蛋白(spike)引发的肥大细胞脱颗粒可诱发肺泡上皮细胞和毛细血管内皮细胞炎症,从而导致肺损伤。气管和支气管是新冠病毒重要的传播位点,这些组织的炎症可能会促进病毒传播。肥大细胞广泛分布于整个呼吸道,因此,在本研究中,我们探究了肥大细胞及其脱颗粒在气管、支气管上皮细胞炎症诱发中的作用。组织病理分析显示,新冠病毒感染的人源化小鼠气管周围肥大细胞积聚并脱颗粒。肥大细胞脱颗粒可引起气管病变,形成乳头状增生。通过对支气管上皮细胞的转录组分析,我们发现肥大细胞脱颗粒显著改变了多种细胞信号通路,特别是导致免疫反应和上调炎症。利用依巴斯汀或氯雷他定可有效抑制支气管上皮细胞炎症因子的诱导,并减轻小鼠的气管损伤。总之,我们的研究证明了肥大细胞及脱颗粒在新冠病毒引起的过度炎症和组织损伤中起到重要作用。我们的研究结果支持使用依巴斯汀或氯雷他定来抑制新冠病毒引发的脱颗粒,从而防止过度炎症引起的组织损伤。

2024, 39(2): 319-330.
doi: 10.1016/j.virs.2024.03.003
收稿日期: 2023-09-07
录用日期: 2024-03-11
出版日期: 2024-03-14
自然发生的HBV前核心区(PC,G1896A)和/或基础核心启动子区(BCP,A1762T/G1764A)突变在慢性HBV感染者特别是HBeAg阴性患者中普遍存在。然而,PC/BCP突变病毒的复制能力仍不明确。本文荟萃分析显示,仅在HBeAg阴性状态下,携带PC突变的患者血清HBV DNA水平高于未携带突变的患者。单独PC突变或联合BCP突变在细胞模型和水压动力注射小鼠模型中均促进了HBV复制。在人肝嵌合小鼠感染模型中,PC联合BCP突变促进病毒复制及肝细胞内核心蛋白的表达。机制上,携带PC突变的preC RNA可表达核心蛋白和P蛋白,这种pgRNA样功能有利于HBeAg阴性状态下cccDNA池的维持。此外,PC联合BCP突变病毒感染在人肝嵌合小鼠模型中可能通过激活内质网应激和TNF信号通路导致更为广泛和严重的人肝细胞损伤。本研究表明,HBeAg阴性患者可能需要定期监测HBV突变并尽早接受抗病毒治疗以延缓疾病进展。

2024, 39(2): 331-334.
doi: 10.1016/j.virs.2023.12.006
收稿日期: 2023-09-14
录用日期: 2023-12-22
出版日期: 2023-12-28
自2022年5月以来,猴痘病毒(MPXV)在世界范围内爆发,严重威胁着人们的公共卫生安全。对病毒关键蛋白的深入研究有助于探索新的抗病毒药物靶点。新月膜(crescent membranes)在MPXV病毒粒子形态发生中起关键作用。该膜由多种病毒蛋白介导形成,这些蛋白被统称为病毒膜组装蛋白(VMAP),其中,A7是最大的VMAP组分。A7由N-端结构域A7N121(含N端121个氨基酸)和C-端结构域A7C组成。本次研究中,我们证明了A7具有磷脂结合能力,并先后解析了A7N121和A7N137的晶体结构。基于对A7N137的结构分析,我们发现连接区域(122-137氨基酸)呈现出明显的构象摆动,导致A7的N端和C端结构域之间产生不同的空间相对取向。随后,通过B-factor分析及分子动力学模拟实验,我们进一步表征了A7连接区域的构象变化特征。最后,基于上述研究结果,我们提出了A7全长的结构模型,并讨论了A7构象变化的潜在生物学意义。总之,该研究扩展了对MPXV A7蛋白的现有认知,并为下一步研究提供了新的方向。

2024, 39(2): 335-337.
doi: 10.1016/j.virs.2024.01.004
收稿日期: 2023-10-25
录用日期: 2024-01-15
出版日期: 2024-01-20
猴痘病毒在包括中国在内的全球范围内的快速传播,凸显制定抗病毒策略的重要意义。由于缺乏猴痘病毒的临床分离株,国内主要使用痘苗病毒对开发的新型抗病毒药物和疫苗进行测试。在此,我们报道了深圳分离的第一株猴痘病毒毒株hMpxV/China/SZ-SZTH42/2023(简称SZTH42)。将来自患者的qPCR阳性皮肤疱疹液接种Vero E6细胞单层,培养72小时内出现典型的噬斑。通过病毒基因组二代测序证实为猴痘病毒。在系统发育上,分离株SZTH42属于分支IIb的C.1谱系,代表了目前世界范围内流行的IIb分支菌株。免疫荧光检测显示分离株SZTH42与市售痘苗病毒诱导的多克隆抗体发生交叉反应。在此基础上,分别开发了用于病毒滴定的焦点形成试验(Focus Forming Assay,FFA)和中和活性评估的和焦点减少中和试验(Focus-Reduction Neutralization Test,FRNT)。通过FFA测定SZTH42的复制动力学,结果显示感染后48小时的病毒滴度最高,释放的细胞外包膜病毒的比例显著低于细胞相关包膜病毒。通过FRNT方法评估了5名猴痘患者的血浆对MPXV SZTH42分离物的中和活性。总之,本研究成功分离了猴痘病毒,为深入研究MPXV的特征以及抗病毒药物和疫苗的开发提供了关键资源和技术平台。

2024, 39(2): 338-342.
doi: 10.1016/j.virs.2024.01.008
收稿日期: 2023-04-14
录用日期: 2024-01-24
出版日期: 2024-02-01
乙型肝炎病毒(HBV)感染是肝细胞癌形成的主要原因,目前仍然严重危害人类健康。乙型肝炎病毒表达一种多功能的X蛋白(HBx),该蛋白不仅仅影响宿主细胞的许多生命活动,还能通过招募DDB1相关的E3泛素连接酶来降解宿主Smc5/6,从而促进HBV感染。尽管大量的研究拓宽了我们对HBx功能的了解,但针对HBx的调控机制还没有充分阐释。在这项研究中,我们发现宿主的E3泛素连接酶TRIM21可以通过对HBx进行非降解性的泛素修饰来抑制HBV的感染和复制。我们发现TRIM21在细胞内的表达水平与HBV的复制呈现负相关的关系。并且,TRIM21可以直接结合HBx蛋白并催化了HBx上非赖氨酸位点的泛素修饰,此种泛素化修饰并不影响HBx的稳定性。更为重要的是,突变了泛素化位点的HBx可以更强地招募DDB1,并促进HBV的复制。这些研究结果为我们更好地理解HBx的调节机制提供了基础,并为针对HBV感染的药物开发提供新思路。

2024, 39(2): 343-346.
doi: 10.1016/j.virs.2024.01.009
收稿日期: 2023-08-31
录用日期: 2024-01-26
出版日期: 2024-02-01
Highlights
1. Full-length genome sequences of previously unreported reassortant swine influenza A(H1N2) virus from domestic pigs in Hong Kong SAR.
2. Phylogenetic analyses revealed close genetic relationship with swine influenza A(H1N2) viruses circulating in Taiwan.
3. Swine influenza A(H1N2) viruses possess potential risk of pig-to-human transmission.
1. Full-length genome sequences of previously unreported reassortant swine influenza A(H1N2) virus from domestic pigs in Hong Kong SAR.
2. Phylogenetic analyses revealed close genetic relationship with swine influenza A(H1N2) viruses circulating in Taiwan.
3. Swine influenza A(H1N2) viruses possess potential risk of pig-to-human transmission.

新型冠状病毒肺炎(COVID-19)于2019年底开始大流行并迅速蔓延,已造成全球数百万人死亡。不同患者对COVID-19的感染严重程度有所不同。基于人群的meta分析有助于理解SARS-CoV-2感染的遗传危险因素与COVID-19严重程度之间的因果关系。为确定影响COVID-19疾病发展的遗传因素,我们在中国人群(n = 632例病例和3021例对照)中进行了全基因组关联研究。在本研究中,我们总结并回顾了世界范围内在22份出版物的COVID-19危重症患者全基因组关联研究中报道的单核苷酸多态性(SNPs),共纳入>167,695例病例和>3,687,897例对照,包括欧洲、美国、南亚、东亚和非洲人群。然后,我们在上述中国COVID-19危重患者人群中验证了这些变异。结果显示,经Bonferroni校正后(P < 0.05/42 = 0.00119),3个基因(DPP9中的rs12610495和靠近 RNU2-47P 和TYRP1的rs4628342、rs1412074、rs1929456、rs1331346和rs10116714)与中国人群显著相关。此外,在已报道的研究和我们的中国人群数据集中,三个SNPs(即FOXP4的rs1886814、GRM5的rs10831496和OAS1的rs10774671)被精确标记。然而,在合并数据的meta分析中,只有FOXP4中的rs1886814与COVID-19的严重程度相关。综上,我们评估了遗传变异与COVID-19严重程度的相关性,并获得了对中国人群中COVID-19预后的见解。

Due to our negligence, the original version of this article, published online on Mar 14, 2023, contained some mistakes in several Figs. In Fig. 2D, the positions of the bands for protein 3A and 3B were incorrectly shifted. This has been modified in corrected Fig.2 as shown below.

39卷第2期 (2024年4月)
ISSN 1674-0769
EISSN 1995-820X
CN 42-1760/Q
主编: 石正丽
影响因子: 5.5*
*源于2022年JCR
GALLERY



会议信息更多+
新闻更多+
- [05/06]《中国病毒学(英文)》期刊编辑部招聘启事
- [01/11]《中国病毒学(英文)》期刊编辑部招聘启事
- [05/07]Q1区!VS最新影响因子5.5!
- [22/02]2022年VS高被引论文奖发布
- [21/10]第十届新生病毒性疾病控制学术研讨会 | 第一轮通知
- [09/09]肝癌细胞中CK1α上调IFNAR1的表达,从而促进I型IFN抑制HBV复制
- [09/09]一种新的干扰素诱导的长非编码RNA ZAP-IT1阻断寨卡病毒在A549细胞中的复制
- [09/09]首发精神分裂症中,驯化的人内源性逆转录病毒W家族包膜蛋白通过降低5-HT4受体的水平激活SK2
- [09/09]发热伴血小板减少综合征病毒L蛋白功能域和保守残基研究为理解病毒RNA转录/复制机制提供新思路
- [09/09]亲环素A结合AKT1并通过介导AKT/mTOR/NF-κB正反馈环路的激活促进EB病毒的致瘤作用 | VS推荐



