Single-cell transcriptomic analyses of HSV-1 reactivation from latently infected tree shrew and mouse trigeminal ganglia reveal differing molecular and cellular processes
Herpes simplex virus type 1 (HSV-1) infects over 70% of the population and establishes lifelong latent infection with periodic reactivation in humans, resulting in various related diseases. However, the molecular and cellular events underlying the transition of HSV-1 from latency to reactivation remain poorly understood. In this study, we used bulk RNA sequencing and single-cell transcriptomic analyses to dissect the cellular and molecular events of HSV-1 latency-reactivation transition in infected trigeminal ganglia (TG) in both mouse and tree shrew infection models. We found that mice exhibited fluctuating host gene responses during the acute phase and relatively quiescent latency, whereas tree shrews displayed a relatively mild acute phase and active latency characteristics. Single-cell analysis revealed that HSV-1 infects TG neuronal subpopulations expressing growth hormone and pituitary hormones. Importantly, we observed that HSV-1 latency in tree shrew TGs exhibited inhibition of cellular autophagy function, while HSV-1 latency in mice was accompanied by the attenuation of monocyte-related immune surveillance. Given that infected cell protein 0 (ICP0) has autophagy inhibitory activity, we further investigated the role of this viral protein in tree shrew models using an ICP0-deficient HSV-1 strain. Notably, the mutant virus could not undergo spontaneous reactivation from latency. These findings support the hypothesis that ICP0 may be essential for spontaneous reactivation by inhibiting autophagy in vivo.
more >>

The role of T cells in influenza infection and vaccination
Influenza virus infections cause significant illness or death every year, becoming a serious health risk. Currently, influenza vaccines mainly induce responses to antibodies against specific strains, but they do not effectively induce effective T cell-mediated immunity. Humoral immunity relies on the production of antibodies that bind to surface proteins (such as hemagglutinin and neuraminidase) to combat the virus. These antibodies envelope the virus to prevent it from invading cells and also label the virus for phagocytic cells to clear. T cell-mediated immunity relies on cytotoxic cells to kill infected cells to combat the virus. Cytotoxic T cells rely on viral proteins on the surface of infected cell to recognize them. This enables the detection of more viral proteins, such as internal proteins like nucleoproteins. A better understanding of the mechanism by which T cells combat influenza is helpful for vaccine development. In this review, we elaborate on the role of T cells in enhancing anti-influenza immune defense. In addition, we explore the possibility that new influenza vaccines can induce such T cell responses.
more >>

Integrated multiplex PCR and metatranscriptomics reveal upper-lower airway microbial landscapes in pediatric respiratory infections
Despite widespread use of multiple PCR, a substantial proportion of pediatric acute respiratory tract infections (ARTIs) lack identifiable pathogens and are classified as unknown etiology. The microbial characteristics and clinical relevance of these cases remain unclear. In this study, we compared the airway microbiomes of PCR-positive and PCR-negative ARTIs and examined their relationships with sampling site and disease severity. A total of 514 hospitalized children with ARTIs were enrolled. Nasopharyngeal swabs (NS) and bronchoalveolar lavage fluid (BALF) samples were tested using a 22-target multiplex PCR panel and subsequently stratified by pathogen status for pooled metatranscriptomic sequencing to profile active microbial communities, viral genotypes, and antibiotic resistance genes. PCR identified common respiratory pathogens in 77.0% of NS and 54.1% of BALF samples. Metatranscriptomic analysis showed that PCR-negative pools displayed markedly lower viral activity and comparatively higher bacterial transcript abundance, with notable enrichment of Pseudomonas. Microbial signatures differed between upper and lower airway samples and across clinical severity, with severe cases demonstrating increased bacterial burden and Pseudomonas enrichment, whereas mild infections exhibited relatively stronger viral signals. Under current thresholds, antibiotic resistance genes were detected in patient pools but not in healthy controls. Overall, PCR-negative pediatric ARTIs exhibited distinct, bacteria-enriched microbial profiles. Integrating metatranscriptomics with PCR enhances pathogen characterization and reveals site- and severity-related microbial patterns that may support diagnostic evaluation and clinical management.
more >>

Current status of dengue fever epidemics and vaccine development
Dengue fever, an acute mosquito-borne infectious disease caused by dengue virus (DENV), is primarily endemic in tropical and subtropical regions. In recent years, the global incidence of dengue has increased dramatically. Since 2023, widespread outbreaks have been reported across numerous countries in the Americas, Asia and Africa. According to the World Health Organization, more than 5 million dengue cases were reported globally in 2023, while the number surged to over 14 million cases with more than 10,000 deaths in 2024—marking the highest global burden ever recorded. A similar upward trend has been observed in China, which experienced its largest dengue outbreak in a decade in 2024, with Guangdong Province accounting for the majority of domestically reported cases. These epidemiological patterns highlight the rapid expansion of dengue transmission, driven by climate change, accelerated urbanization and increased human mobility. In this context, vaccine development has become a public health priority. To date, two vaccines—Dengvaxia and Qdenga—have been licensed for clinical use. Six other vaccine candidates are currently in clinical trials, among which the tetravalent live-attenuated vaccines TV003/TV005 are considered the most promising. Despite considerable advances in dengue vaccine research, significant challenges remain, including the need to elicit balanced immune responses against the four serotypes and to reduce the risk of antibody-dependent enhancement (ADE). Taken together, this review systematically summarizes recent global and regional trends in dengue fever and the current progress in dengue vaccine development, collectively offering a valuable resource for informing prevention and control strategies.
more >>

Multivalent display of envelope protein domain III with Mi3 nanoparticles induces protective immunity against lethal Zika virus infection in mice
Zika virus (ZIKV) infection is associated with severe neurological complications such as congenital microcephaly, yet no safe and effective vaccine is currently available. A critical challenge in ZIKV vaccine development arises from cross-reactive, non- or sub-neutralizing antibodies, which may enhance dengue virus (DENV) infection through antibody-dependent enhancement (ADE). Herein, we report a vaccine strategy utilizing Mi3 nanoparticles to display the envelope (E) protein domain III (EDIII) of ZIKV, which induces protective immunity against ZIKV infection in murine models. Compared to an EDIII subunit vaccine, the Mi3-EDIII nanoparticle vaccine elicited significantly higher antibody responses and stronger cell-mediated immune responses. In C57BL/6 mice, maternal immunization with Mi3-EDIII protected the neonates against ZIKV-caused symptoms, including body weight loss, neurological abnormalities, retardation of brain development, and mortality. In interferon-α/β receptor knockout (Ifnar1-/-) C57BL/6 mice, Mi3-EDIII immunization conferred effective protection against lethal ZIKV challenge. Notably, unlike ZIKV convalescent sera, Mi3-EDIII immune sera did not enhance DENV infection in human chronic myelogenous leukemia K562 cells, suggesting the absence of ADE-prone antibody induction. Our results demonstrate that Mi3-EDIII is a promising vaccine candidate against ZIKV infection and warrants further development.
more >>

Metatranscriptomics profiling reveals rodent- and shrew-borne viral diversity and evolutionary relationships in Guangzhou, China
Emerging zoonotic infectious diseases, predominantly caused by viruses, pose increasing public health threats globally. Rodents and shrews are natural hosts for a variety of zoonotic viruses. Guangzhou is one of China’s most densely populated cities and experiences frequent international and domestic population movements, making it a hotspot for infectious diseases. This study reports the metatranscriptomics virome of 208 rodents and shrews collected between June 2023 and December 2024 from four main urban areas (Tianhe, Baiyun, Liwan, Yuexiu) and five non-main urban areas (Zengcheng, Huadu, Conghua, Panyu, Nansha) in Guangzhou. Individual libraries were constructed from mixed tissue samples (liver, spleen, lung, and kidney) of each animal. Metatranscriptomics sequencing revealed diverse viral communities, identifying 24 viral strains across eight mammalian-associated viral families. Notably, we identified 17 known viruses and seven potentially novel viruses, including Seoul virus (5.2% prevalence in Rattus norvegicus from Panyu), Wenzhou mammarenavirus (13.2% in Rattus norvegicus from Conghua and Huadu), Jeilongvirus (29.4% in Rattus andamanensis from Panyu), and a divergent lineage of arteriviruses that may represent a new genus (maximum positivity rates of 2.9% in Rattus norvegicus and 5.7% in Rattus tanezumi). Phylogenetic analysis elucidated evolutionary relationships within key families such as Hantaviridae, Arenaviridae, Flaviviridae, and Parvoviridae, revealing distinct viral carriage patterns in Guangzhou City that are shaped by host species and geographical location. This is the first macro-level study of rodent and shrew viromes in Guangzhou and provides a scientific basis for strengthening surveillance of mammalian-associated viruses and preventing emerging zoonotic infectious diseases in the region.
more >>
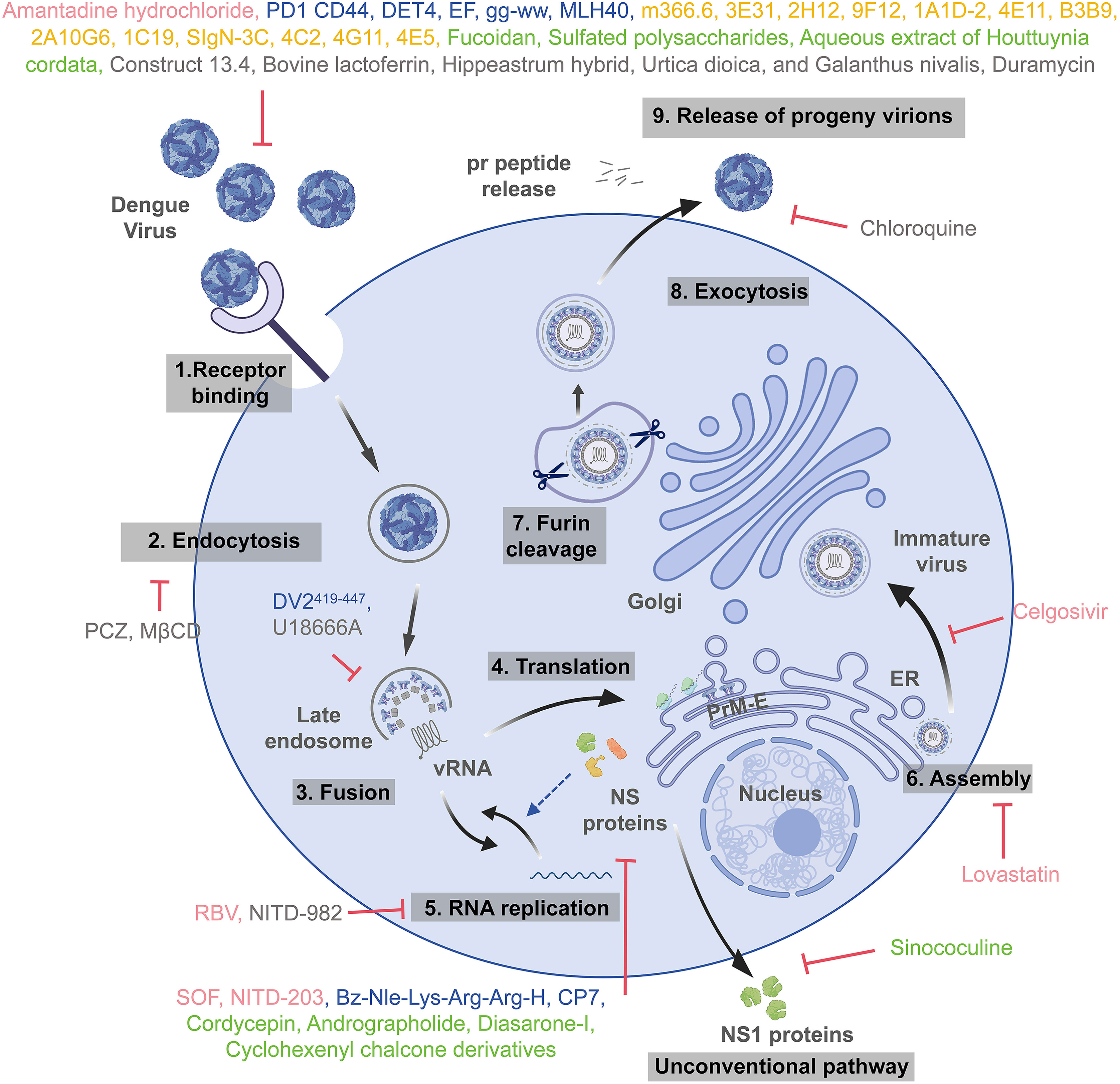
Antiviral agents for dengue virus
Dengue virus (DENV) is a mosquito-borne pathogen responsible for a spectrum of illnesses, including dengue fever, dengue hemorrhagic fever, and dengue shock syndrome. Nearly half of the global population is at risk of DENV infection, making it a pressing public health issue worldwide. The limited cross-protection among the four DENV serotypes (DENV1-4) and the phenomenon of antibody-dependent enhancement (ADE) have posed significant challenges to the development of effective dengue vaccines. Furthermore, there are currently no specific antiviral treatments available. This review provides an overview of DENV's key characteristics, clinical manifestations, and recent advancements in antiviral drug development—including the repurposing of approved drugs, peptide-based antiviral agents, therapeutic antibodies, natural products with antiviral potential, and host factor inhibitors—aiming to offer critical insights to inform strategies for managing and preventing dengue outbreaks.
more >>

The coronavirus 3CL protease: Unveiling its complex host interactions and central role in viral pathogenesis
The 3CL protease, a highly conserved enzyme in the coronavirus, plays a crucial role in the viral life cycle by facilitating viral replication through precise cleavage of polyproteins. Beyond its proteolytic function, the 3CL protease also engages in intricate interactions with host cell proteins involved in critical cellular processes such as transcription, translation, and nuclear-cytoplasmic transport, effectively hijacking cellular machinery to promote viral replication. Additionally, it disrupts innate immune signaling pathways, suppresses interferon activity and cleaves antiviral proteins. Furthermore, it modulates host cell death pathways including pyroptosis and apoptosis, interferes with autophagy and inhibits stress granule formation to maintain viral infection and exacerbate viral pathogenesis. This review highlights the molecular mechanisms by which the 3CL protease orchestrates virus-host interactions, emphasizing its central role in coronavirus pathogenesis and highlighting potential therapeutic targets for future interventions.
more >>

Virome diversity in small mammals from south China: Insights into virus evolution, transmission, and ecology
Mammals are critical reservoirs of human infectious diseases and the spillover of viruses is related to climate conditions. We conducted meta-transcriptomic sequencing of 226 mammals (bats, rodents, hedgehogs, and shrews) representing 20 species collected across eight cities in south China between 2018 and 2024. Samples included internal organs, oropharyngeal and anal swabs, and feces. We identified 63 vertebrate-associated viruses, including 34 novel viruses. Phylogenetic analysis revealed six viruses with potential infection risks to humans or domestic animals due to their close phylogenetic relationships with known pathogens. Cross-species transmission was observed in 14.3% (9/63) of viruses, shared by at least two host species, with bats, particularly Rhinolophus and Hipposideros, serving as key hubs for viral circulation and zoonotic spillover. Virome composition varied substantially among mammalian species and geographic regions (adonis test, R2 = 0.50, P = 0.001). Generalized linear models quantified the roles of host taxonomy, ecotypes, and meteorological factors in shaping viral diversity, demonstrating host taxonomy (at the order level) as a predominant role (25.70% deviance explained), followed by ecotypes (10.27% deviance explained). Phylogenetic analysis conducted using our betacoronavirus sequences, as well as betacoronavirus sequences derived from 2.0 × 104 bats sampled in China between July 2013 and March 2024, revealed that no betacoronaviruses exhibited closer phylogenetic relationships to SARS-CoV-2 than the known strains (e.g., RaTG13). These findings provide critical insights into virus evolution, transmission, and ecological determinants, which are essential for the prevention of emerging infectious diseases.
more >>

Host factor RBM25 promotes HBV replication through Yin Yang 1-mediated cccDNA transcription
The persistence of covalently closed circular DNA (cccDNA) in hepatitis B virus (HBV)-infected hepatocytes remains a major obstacle to effective antiviral treatment. Understanding the molecular mechanisms regulating HBV cccDNA transcription is essential for developing novel therapeutic strategies. In this study, we investigated the role of RNA binding motif protein 25 (RBM25) in HBV replication, focusing on its interaction with cccDNA and its regulation of host transcription factors. The results demonstrated that RBM25 knockdown markedly inhibited HBV replication, reducing levels of HBV DNA, hepatitis B e antigen (HBeAg), hepatitis B surface antigen (HBsAg), HBV RNA, and L-HBs in HBV-replicating and infected cell models. Consistent results were observed in a mouse model hydrodynamically injected with 1.2 × HBV plasmid. Conversely, RBM25 overexpression significantly enhanced HBV replication. Mechanistically, RBM25 promoted HBV promoter activities by binding to cccDNA through its RE/RD and PWI domains. This effect was mediated by increased Yin Yang 1 (YY1) expression, which enhanced acetylation of cccDNA-bound histones, promoting HBV transcription. Furthermore, RBM25 expression was upregulated and translocated to the nucleus following core protein expression and accumulation, while overexpression of RBM25 promoted core protein degradation. In conclusion, this study demonstrates that RBM25 is a novel host factor that enhances HBV replication by upregulating YY1-dependent transcriptional activation of cccDNA. It also reveales a reciprocal regulatory mechanism between the HBV core protein and RBM25, which helps sustain HBV replication.
more >>

Insights into cross-species infection: Porcine epidemic diarrhea virus infections in the rodent
The cross-species infection of coronaviruses has resulted in several major epidemics since 2003. Porcine epidemic diarrhea virus (PEDV) is a devastating swine enteric coronavirus, which targets pigs as the only natural reservoir. In this study, the nucleic acid of PEDV was detected in rat fecal samples collected from pig farms. Further animal tests showed that PEDV can cause systemic infections in neonatal mice and rats via intracranial inoculation. The brain, lung, intestine and spleen were all targets for PEDV in rodents in contrast to the intestine being targeted in pigs. Morbidity and mortality vary via different infection routes. PEDV was also detectable in feces after infection, suggesting that the infected rodents were potential infectious sources. Moreover, the cerebral tropism of PEDV was verified in piglets via orally inoculation, which had not been identified before. In conclusion, our findings demonstrate that PEDV could cross the species barrier to infect mice and rats through different routes in experimental settings. Although it is highly devastating to piglets, PEDV changes the target organs and turns to be milder when meeting with new hosts. Based on these findings, more attention should be paid to the potential cross-species infection of PEDV.
more >>

Qingqi Guxue Decoction induces S cell cycle arrest to inhibit replication of severe fever with thrombocytopenia syndrome virus
Severe fever with thrombocytopenia syndrome (SFTS) is a novel emerging acute infectious disease caused by severe fever with thrombocytopenia syndrome virus (SFTSV), characterized by high fever and thrombocytopenia. It has been proved that traditional Chinese medicine (TCM) has displayed definite therapeutic effects on viral hemorrhagic fever, indicating its potential to treat SFTS. In this study, SFTS-relative key targets were predicted via gene ontology (GO) analysis and kyoto encyclopedia of genes and genomes (KEGG) enrichment analysis. Molecular docking was then used to select stable binders. Molecules matched TCMs were identified, and a new prescription, Qingqi Guxue decoction (QQGX), was formulated to clear heat and nourish blood, with a resulting drug composition network. We explored the optimal drug proportion for QQGX. Through an in-depth study of molecular mechanisms, we found that QQGX induces S phase arrest by promoting the degradation of cyclin A2 (CCNA2) and cyclin-dependent kinase 2 (CDK2), thereby inhibiting SFTSV replication. Finally, we verified the effectiveness and safety of QQGX based on the mouse liver bile duct organoid model infected with SFTSV. In summary, our study prepared a TCM decoction using the method of network pharmacology. This decoction has a significant inhibitory effect on the replication of SFTSV and provides a new treatment strategy for hemorrhagic fever with TCM.
more >>

Zika virus transmission in Aedes aegypti: A systematic study on the ability of mosquitoes to transmit the virus horizontally and vertically
Zika virus (ZIKV) is a mosquito-borne virus belonging to the genus Orthoflavivirus, and the family Flaviviridae. It commonly presents with febrile-like symptoms, neurological issues, and pregnancy complications in humans. Currently, there is no commercial vaccine or specific treatment available to prevent ZIKV infection. Therefore, controlling the epidemic's spread relies on preventing mosquitoes from transmitting the virus. Although various studies have explored the transmission of ZIKV between mosquitoes and vertebrate hosts, comprehensive research on potential mosquito-to-mosquito transmission of ZIKV remains limited. In this study, we conducted systematic laboratory investigations to assess the ability of ZIKV to spread among mosquitoes, and to evaluate the impact of ZIKV infection on mosquito development. Our findings revealed that ZIKV can be transmitted between Aedes aegypti mosquitoes both vertically and horizontally, through oviposition and contact between mosquitoes of the same or opposite sex. Additionally, we observed that ZIKV infection resulted in a reduction in the number of mosquito eggs but an increase in their size. The widespread distribution of ZIKV in infected mosquitoes and the altered levels of hormone related genes following viral infection were noted, which may contribute to viral transmission among mosquitoes and affect mosquito development. This research provides systematic experimental evidence of ZIKV transmission among mosquitoes, which is crucial for developing novel strategies to disrupt the spread of orthoflaviviruses and other mosquito-borne pathogens.
more >>

Current antiviral therapies and promising drug candidates against respiratory syncytial virus infection
Respiratory syncytial virus (RSV) is one of the most common viruses leading to lower respiratory tract infections (LRTIs) in children and elderly individuals worldwide. Although significant progress in the prevention and treatment of RSV infection was made in 2023, with two anti-RSV vaccines and one monoclonal antibody approved by the FDA, there is still a lack of postinfection therapeutic drugs in clinical practice, especially for the pediatric population. In recent years, with an increasing understanding of the pathogenic mechanisms of RSV, drugs and drug candidates, have shown great potential for clinical application. In this review, we categorize and discuss promising anti-RSV drug candidates that have been in preclinical or clinical development over the last five years.
more >>

Insights into recent advancements in human and animal rotavirus vaccines: Exploring new frontiers
Rotavirus infections cause severe gastroenteritis and dehydration in young children and animals worldwide, leading to high rates of morbidity and mortality, predominantly in low- and middle-income countries. In the past decade, substantial progress has been made in the development and implementation of rotavirus vaccines, which have been essential in alleviating the global burden of this disease, not only in human being but also in livestock species like calves and piglets, where these infections can cause significant economic losses. By synthesizing the latest research and real-world evidence, this review article is designated to provide deep insights into the current state of rotavirus vaccine technology and its global implementation as well as the application of rotavirus vaccines in veterinary settings and their importance in controlling zoonotic transmission and maintaining food security.
more >>
Articles in press have been peer-reviewed and accepted, which are not yet assigned to volumes/issues, but are citable by Digital Object Identifier (DOI).
|
2026, 41(3): 493-512.
doi: 10.1016/j.virs.2025.12.006
Received: 01 April 2025
Accepted: 04 December 2025
Published: 15 December 2025


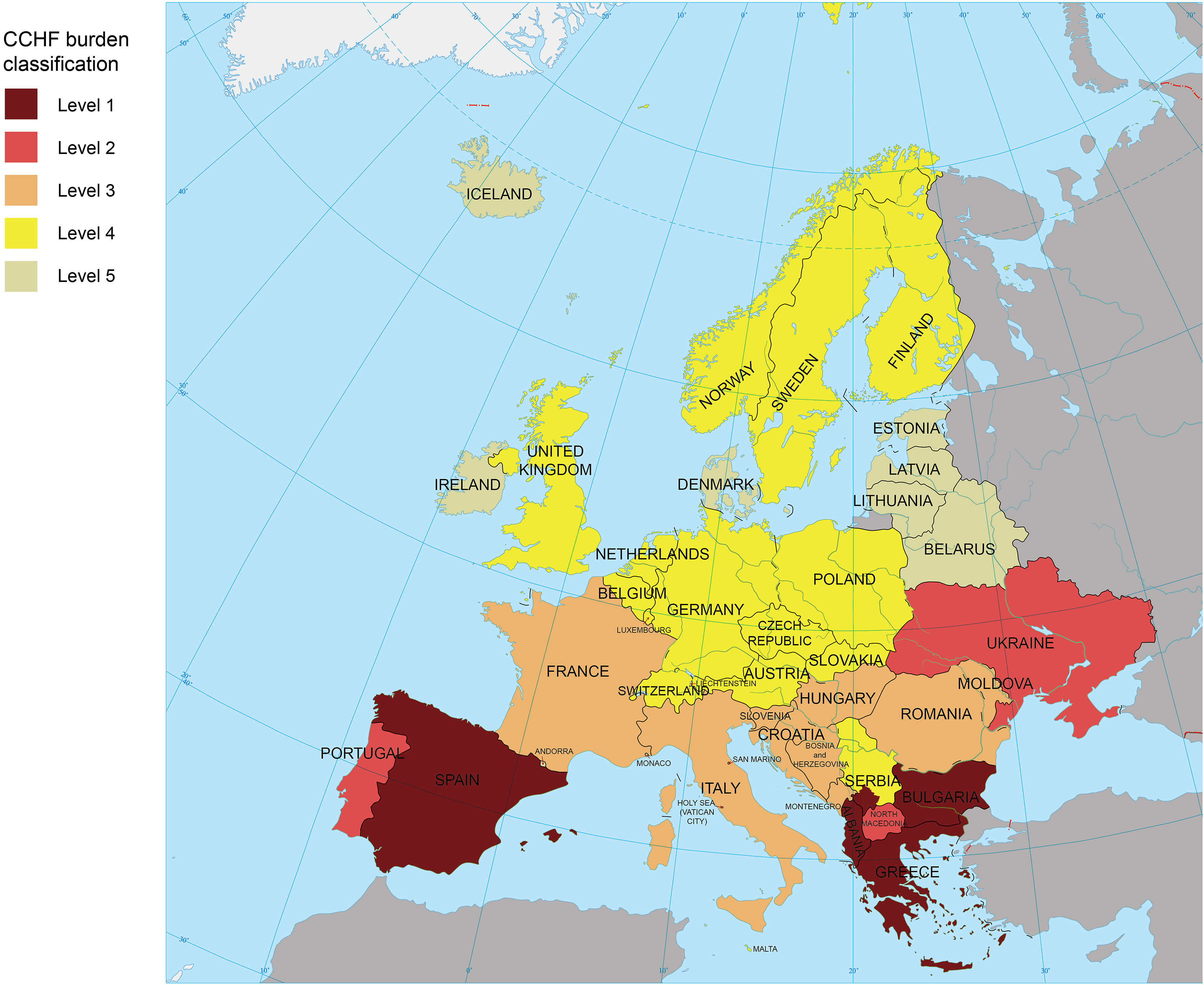
Crimean-Congo hemorrhagic fever (CCHF), caused by Crimean-Congo hemorrhagic fever virus (CCHFV), is endemic in Africa, Asia, and Europe. However, CCHF epidemiology and epizootiology have been poorly defined in Europe. Here, we summarize the current knowledge of CCHFV distribution in (non-Russian) Europe, including countries previously not considered to be at risk. We collected data on CCHF cases, human/vertebrate animal anti-CCHFV seroprevalence, CCHFV vector (Hyalomma tick), and CCHFV isolation from ticks and classified countries into five risk levels using a One Health approach. From 1944 through Feb 2025, more than 2000 recorded CCHF cases were identified in Europe, mostly from southern/eastern countries/regions, primarily Bulgaria (at least 1623), Kosovo (at least 339), Ukraine (at least 336), Croatia (at least 200), Albania (at least 146), and Republic of Moldova (at least 60). Albania, Bulgaria, Greece, Kosovo, and Spain were categorized as level 1 (reported CCHF cases, presence of robust surveillance systems). North Macedonia, Portugal, and Ukraine/Crimea were assigned to level 2 (reported CCHF cases in the absence of robust established surveillance). Bosnia and Herzegovina, Croatia, France, Hungary, Italy, Montenegro, Republic of Moldova, Romania, and Slovenia were assigned to level 3 due to evidence of CCHFV circulation in absence of recent CCHF cases. Thirty-four countries were assigned to level 4 (presence of Hyalomma ticks) or level 5 (no data). This work provides information on CCHFV distribution and burden with list of at-risk areas to inform international and local public health agencies to establish or strengthen surveillance systems.
2026, 41(3): 513-522.
doi: 10.1016/j.virs.2026.05.011
Received: 05 September 2025
Accepted: 29 May 2026
Published: 03 June 2026
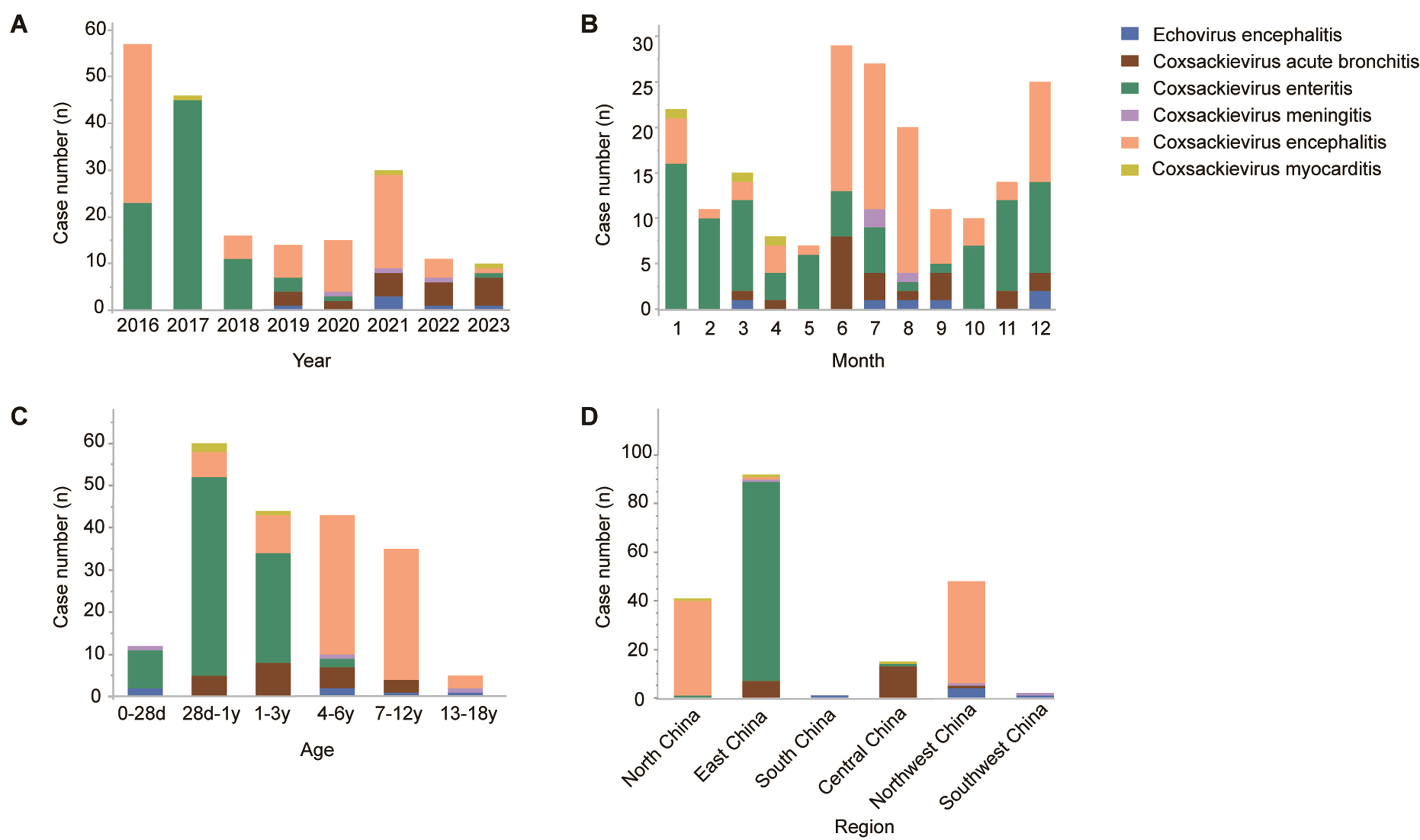
Enterovirus (EV) infections represent a significant global and national health concern in children, leading to various complications. Although most children with EV infections generally have a favorable prognosis, a small proportion may still develop severe complications. This study aimed to analyze the epidemiological characteristics, disease spectrum, and disease burden of EV infections in China. A total of 163,714 hospitalized children with EV infections from 37 member hospitals of the Futang Research Center of Pediatric Development were identified between Jan 1st, 2016 and Dec 31st, 2023, accounting for 1.49% of all pediatric hospitalizations. Most cases occurred in infants aged 28 days to ≤ 1 year (42.29%) and toddlers aged 1 to ≤ 3 years (39.93%), with a male predominance (male-to-female ratio, 1.62 : 1). Severe cases of EV infections can be life-threatening and increase the burden of disease. The median LOS for EV infections were 5 days, with an average hospitalization cost of $587.37. EV infections can affect multiple organs, including the heart, brain, and respiratory system and were associated with severe complications, including myocarditis, encephalitis, and meningitis. These findings underscore the substantial disease burden of EV infections and highlight the need for targeted prevention strategies in young children.
2026, 41(3): 523-531.
doi: 10.1016/j.virs.2026.06.001
Received: 12 December 2025
Accepted: 29 May 2026
Published: 03 June 2026
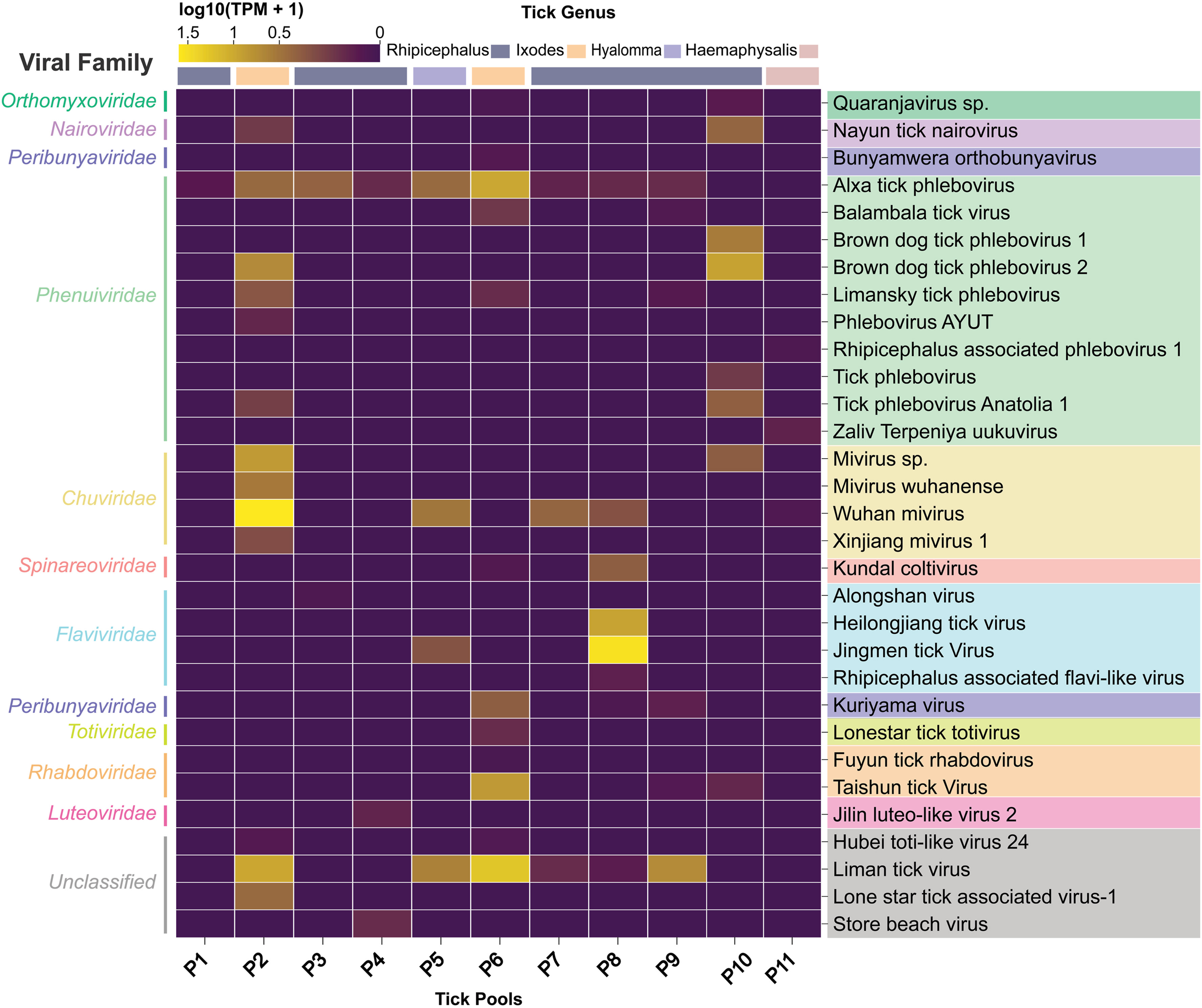
Tick-borne viruses (TBVs) pose significant emerging threats to public and veterinary health worldwide. In Pakistan, the potential threats posed by TBVs extend far beyond Crimean-Congo hemorrhagic fever virus (CCHFV), which causes outbreaks and severe hemorrhaging with a high fatality rate among humans each year. However, the full extent of the tick-borne virome remains largely unexplored. This study presents the metagenomic profiling of viruses in livestock-associated ticks from Pakistan. Eighty-seven ticks belonging to the genera Ixodes, Rhipicephalus, Haemaphysalis, and Hyalomma species from livestock in Punjab. These ticks were subsequently grouped into 11 pools for RNA sequencing. Our analysis revealed extensive viral diversity, identifying sequences related to 31 viruses spanning at least 11 families. New strains of Jingmen tick virus (JMTV), Brown dog tick phlebovirus 2 (BDTPV-2), and Liman tick virus (LMTV) were characterized, confirming their presence in the region. Serological surveys performed among 319 livestock, 253 humans, and 214 rats detected antibodies against these viruses, indicating host exposure. Notably, the presence of JMTV-neutralizing antibodies was confirmed in two livestock animals, one human, and one rat, providing evidence of productive infection. Our findings significantly expand the known diversity and distribution of TBVs in Pakistan, establish the preliminary baseline of the tick virome in the country, and provide serological evidence of cross-species exposure to emerging TBVs. This study highlights the underestimated risk of tick-borne viral zoonoses in Pakistan and underscores the urgent need for enhanced surveillance and risk assessment.
2026, 41(3): 532-542.
doi: 10.1016/j.virs.2026.05.009
Received: 22 February 2026
Accepted: 27 May 2026
Published: 03 June 2026
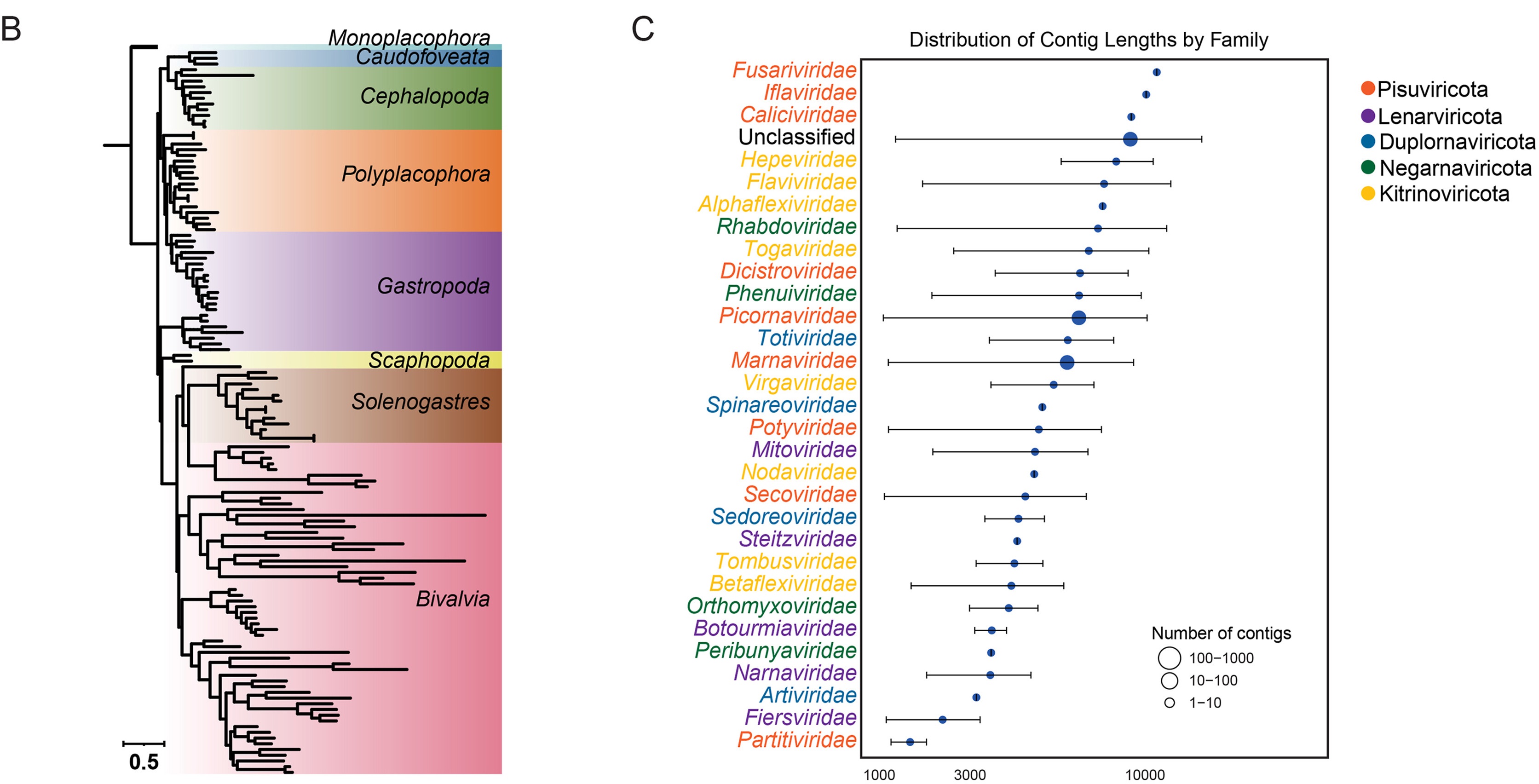
Mollusca, the second-largest animal phylum, includes many aquaculture species used as important food resources for humans. While DNA viruses that threaten molluscan aquaculture have received much attention, molluscan RNA viromes are still understudied. Here, using a multi-stage RdRP discovery pipeline combining six-frame translation, profile-based homology search, and phylogenetic validation, 203 RNA viruses spanning five viral phyla and fifteen viral orders were identified, based on 223 molluscan metatranscriptomes covering eight classes. Phylogenetic analysis, combined with structural modeling, revealed a significant increase in the number of Pisuviricota-related lineages. Extensive modular evolution in viral genomes was observed, including gene rearrangements and co-evolution of capsid and RdRP genes. Host prediction linked 76% of the RNA viruses to a range of eukaryotes. These findings expand the diversity of RNA viruses associated with molluscs and shed light on their phylogenetic relationships, highlighting that molluscs might serve as an important reservoir of diverse RNA viruses infecting a range of eukaryotic hosts.
2026, 41(3): 543-560.
doi: 10.1016/j.virs.2026.03.001
Received: 03 September 2025
Accepted: 06 March 2026
Published: 12 March 2026
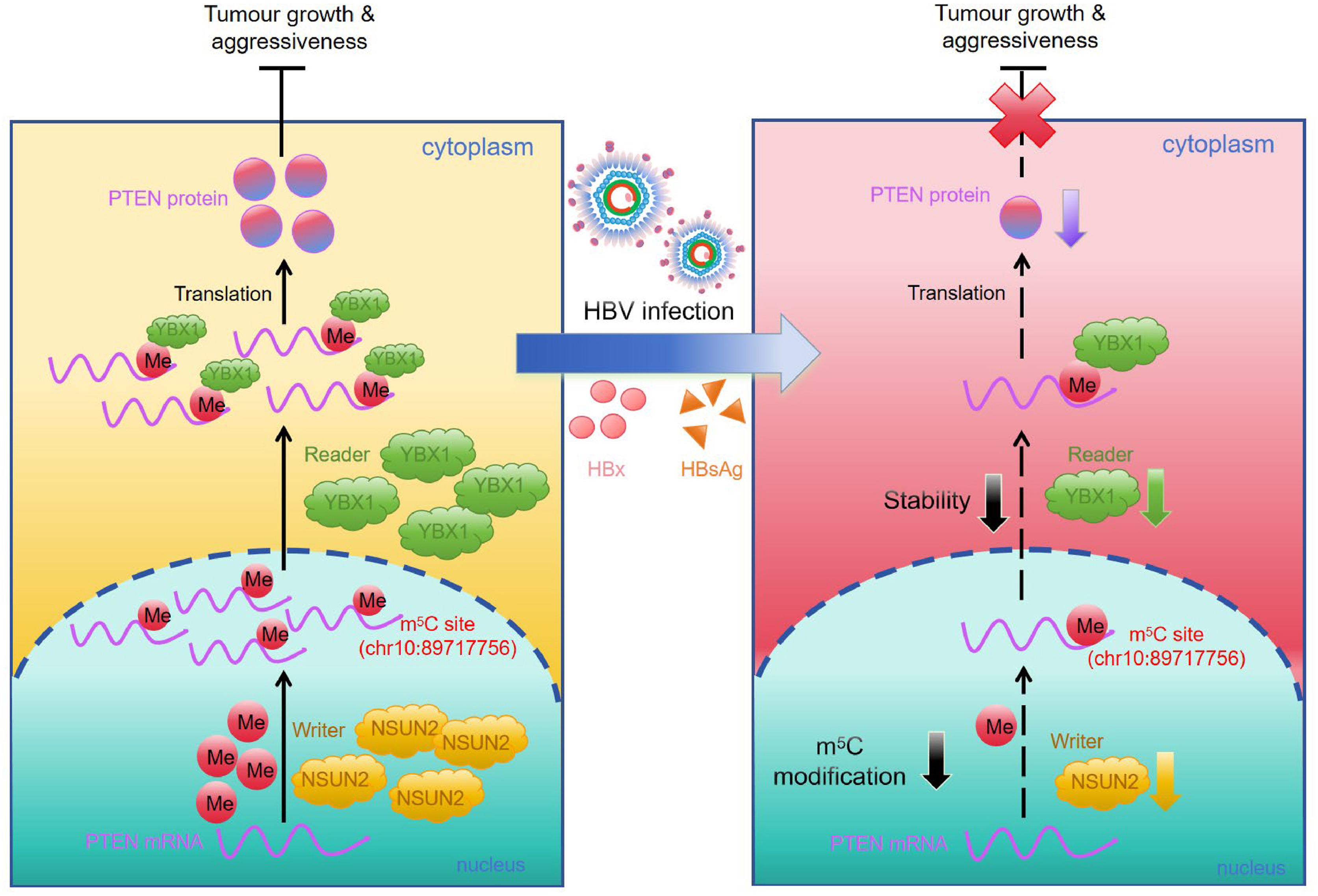
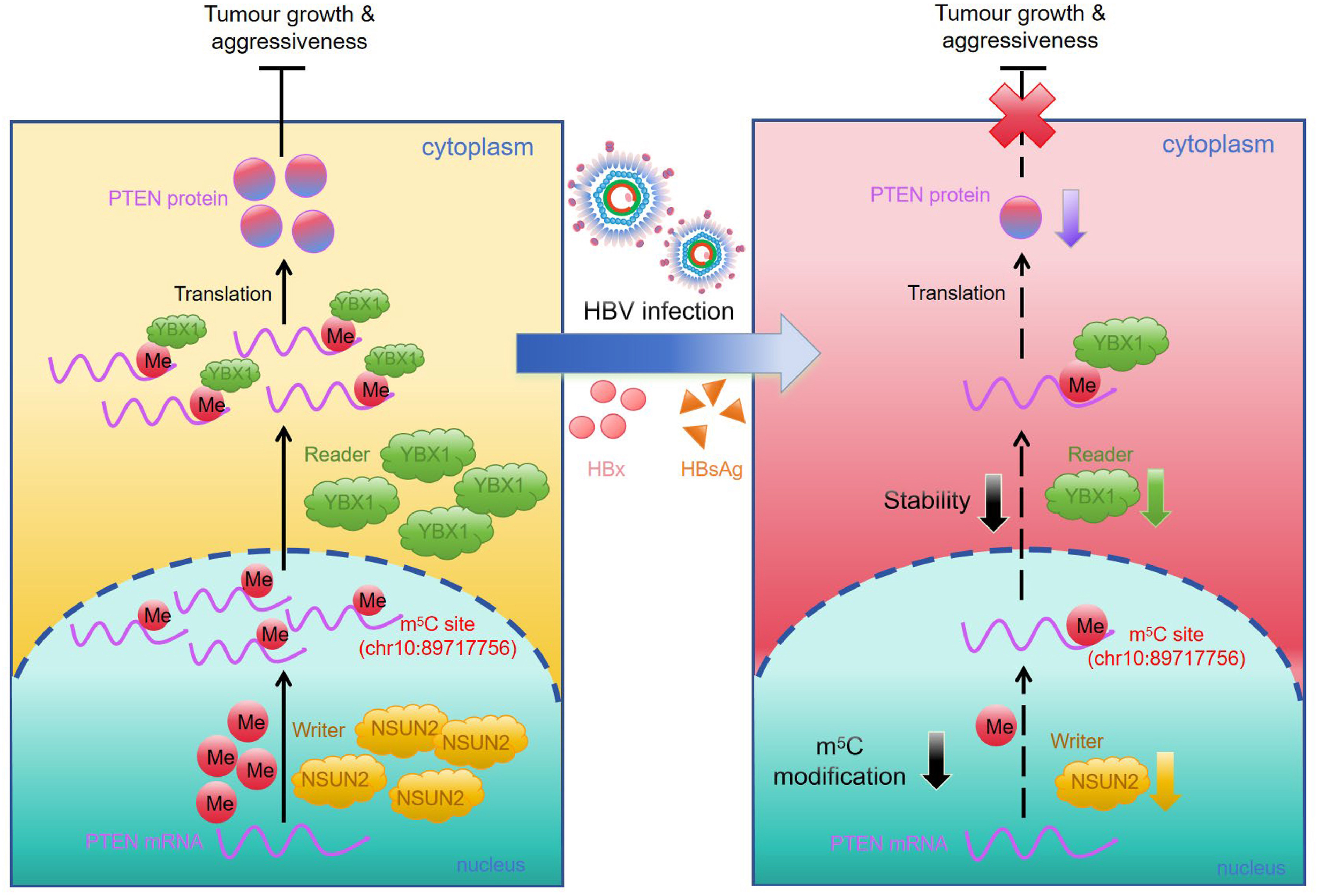
Hepatitis B virus (HBV) has been implicated in hepatocellular carcinoma (HCC) progression, partly through regulation of the tumor suppressor phosphatase and tensin homolog (PTEN). Although transcriptional regulation of PTEN by HBV is well characterized, its post-transcriptional regulation remains poorly understood. Because RNA 5-methylcytosine (m5C) modification influences post-transcriptional gene control and cancer development, we investigated whether HBV modulates PTEN through m5C. Methylated RNA immunoprecipitation (MeRIP)-quantitative polymerase chain reaction showed a marked reduction in m5C on PTEN mRNA in HBV-producing cells. MeRIP sequencing further identified decreased m5C within the PTEN coding sequence region (chr10:89717747-89717771) in HBV-producing HepAD38/tetracycline-off cells, with chr10:89717756 emerging as a critical site where HBV suppresses m5C enrichment and PTEN expression. Mechanistically, the m5C “writer” NOP2/Sun RNA methyltransferase 2 (NSUN2) and the “reader” Y-box binding protein 1 (YBX1) stabilized PTEN mRNA in an m5C-dependent manner. HBV disrupted this pathway, decreasing PTEN mRNA stability via NSUN2- and YBX1-mediated m5C. Overexpression of NSUN2 or YBX1 attenuated HBV-driven proliferation, migration, and invasion, and these effects were partially reversed by the PTEN inhibitor VO-Ohpic. The small hepatitis B surface antigen and hepatitis B X protein downregulated NSUN2 and YBX1, linking viral proteins to PTEN suppression. Further, HBV is associated with reduced NSUN2 expression in HBV transgenic (HBV-Tg) mice, HBV-infected primary human hepatocytes as well as HBV-positive clinical HCC specimens, supporting the physiological and clinical relevance of this finding. Together, these findings identify the NSUN2/YBX1/PTEN axis as a potential therapeutic target in HBV-associated HCC.
2026, 41(3): 561-573.
doi: 10.1016/j.virs.2026.05.001
Received: 11 January 2026
Accepted: 09 May 2026
Published: 14 May 2026
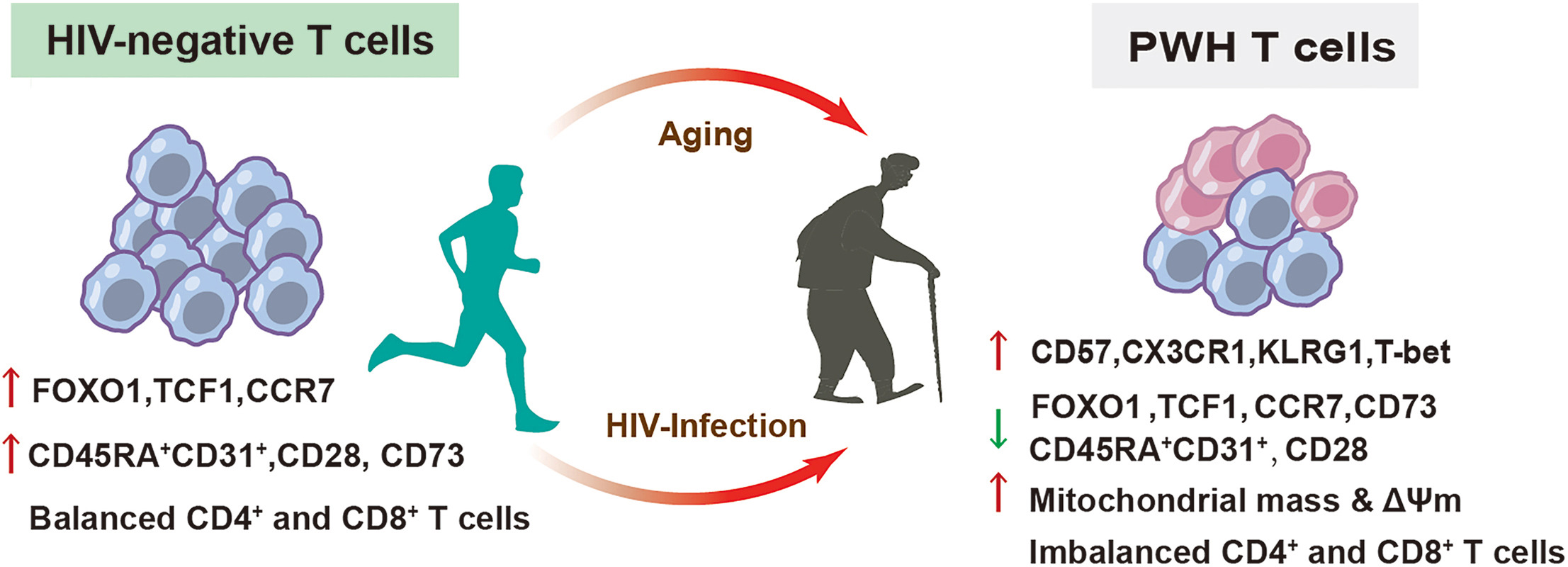
Antiretroviral therapy (ART) has significantly extended the life expectancy of people with HIV (PWH), rendering population ageing and immunosenescence prominent clinical priorities. T-cell senescence is linked to mitochondrial dysfunction and drives age-related immune remodelling, yet how HIV infection and ageing jointly shape CD4+ and CD8+ T-cell immunophenotypes and mitochondrial remodelling remains unclear. This cross-sectional study included 61 PWH on suppressive ART for ≥ 12 months and 61 age- and sex-matched HIV-negative men who have sex with men, stratified into younger (≤ 35 years) and older (≥ 50 years) groups. Multiparameter flow cytometry was used to profile CD4+ and CD8+ T-cell differentiation, stemness, activation/exhaustion, and metabolic phenotypes, together with mitochondrial mass and membrane potential. We found that ageing and HIV infection were associated with T-cell remodelling, characterized by expanded late-differentiated phenotypes and reduced stem-like, homeostatic and costimulatory CD4+ and CD8+ T-cell subsets, as indicated by upregulated CD57 and CX3CR1 and downregulated CD45RA+CD31+, FOXO1, and CD28. Notably, younger PWH had an ageing-like CD4+ T-cell profile, with higher CD57, CX3CR1 and TIGIT expression than younger HIV-negative individuals. In contrast, HIV-related CD8+ T-cell perturbations (KLRG1, CXCR3, NKG2C and CD95) were more pronounced in older PWH. PWH exhibited increased mitochondrial mass and membrane potential in both total and senescent-like CD4+ and CD8+ T cells, particularly in CD8+ T cells from older PWH. In CD4+ T cells, KLRG1 and CX3CR1 expression correlated positively with age, and inversely with CD4+ T-cell counts and CD4/CD8 ratio. By contrast, FOXO1 expression in CD8+ T cells was inversely associated with age, late-differentiation markers, and ART duration in PWH. Overall, age is a major driver of T-cell immunosenescence, and HIV infection modulates and exacerbates these alterations. Mitochondrial stress, FOXO1 downregulation and immune network remodelling support a multifaceted model of HIV-associated immune ageing that may contribute to heterogeneous immune reconstitution in PWH, highlighting potential targets to mitigate immune ageing.
2026, 41(3): 574-585.
doi: 10.1016/j.virs.2026.05.002
Received: 07 January 2026
Accepted: 12 May 2026
Published: 14 May 2026
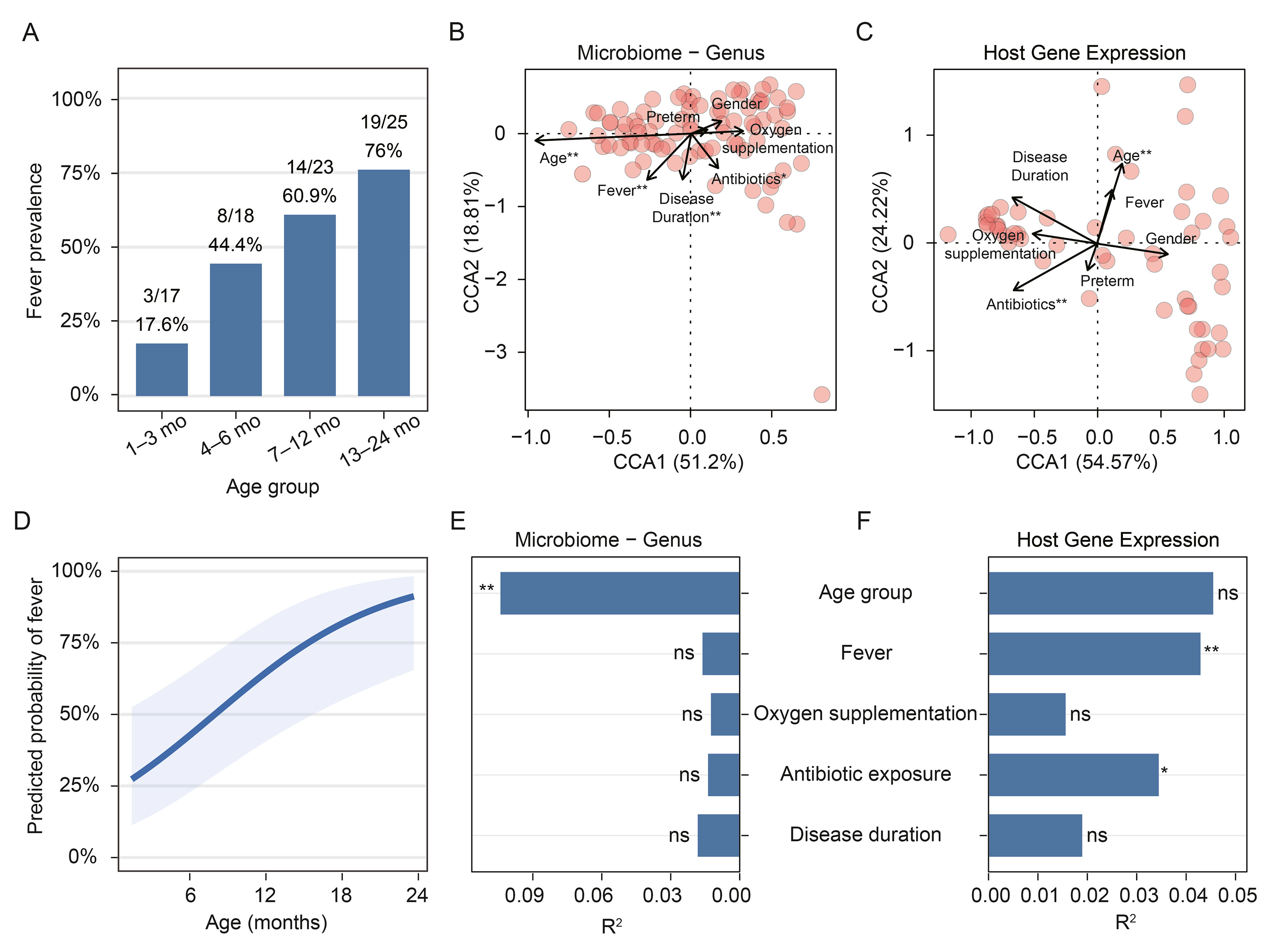
Respiratory syncytial virus (RSV) bronchiolitis is the leading cause of hospitalization in infancy and exhibits pronounced age-dependent clinical heterogeneity. Fever becomes increasingly prevalent with age, yet whether febrile representation reflects a uniform inflammatory and immune phenotype across infancy remains unclear. In this prospective cohort of infants hospitalized with RSV bronchiolitis, we performed an integrated analysis of clinical features, pharyngeal microbiome composition, host transcriptomic profiles, and host-microbe interaction networks, with particular attention to age-related variation in fever-associated patterns. Clinically, fever prevalence exhibited a strong age-dependent increase across infancy. Correspondingly, canonical correspondence analysis identified age and fever as dominant gradients related to variation in both pharyngeal microbiome composition and host gene expression. Although no significant age-dependent correlations were observed at the global microbial and host transcriptomic levels in the fever-age interaction model, distinct patterns of microbial and host responses related to fever were observed across different age groups. Specifically, ranked gene set enrichment analysis indicated that febrile infants in early infancy showed relative attenuation of host defense-related programs, whereas older infants showed stronger enrichment of antiviral and inflammatory effector pathways, with more selective regulatory and signaling-associated patterns in late infancy. Integrated host-microbe network analysis further delineated a coherent developmental trajectory of fever-associated interaction architectures, evolving from densely interconnected regulatory networks in early infancy to modular, selectively coupled, host-centered configurations with advancing age. Together, febrile responses in RSV bronchiolitis should not be interpreted as a uniform biological phenotype across infancy and support age-aware interpretation of fever in pediatric RSV infection.
2026, 41(3): 586-601.
doi: 10.1016/j.virs.2026.05.004
Received: 12 November 2025
Accepted: 21 May 2026
Published: 27 May 2026
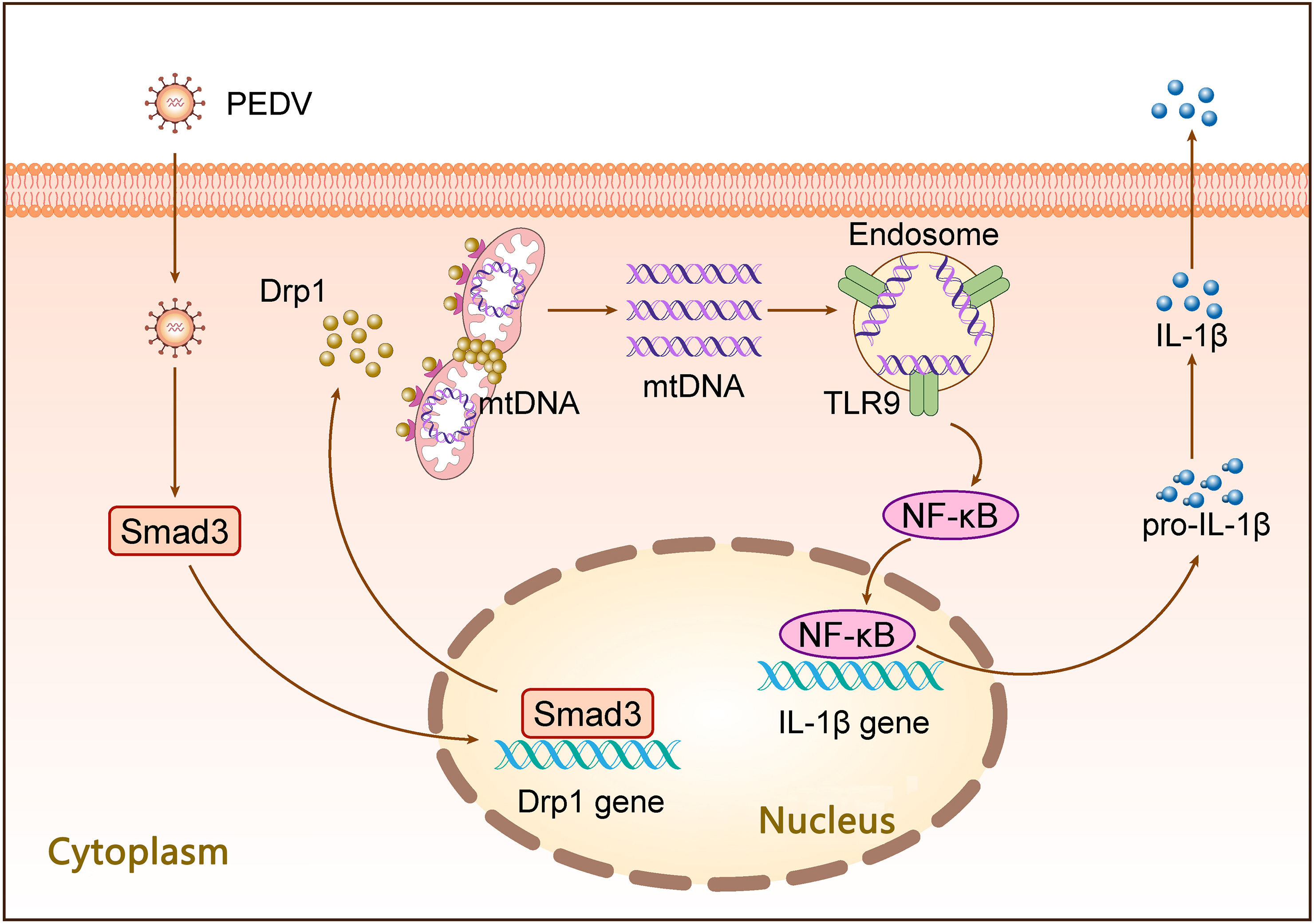
Mitochondrial homeostasis is intricately linked to the pathogenesis of many viral infections. The maintenance of mitochondrial homeostasis and normal cellular functions relies heavily on the delicate balance of mitochondrial dynamics. However, the precise impact of porcine epidemic diarrhea virus (PEDV) infection on mitochondrial dynamics and subsequent pathological processes is largely unexplored. In this study, through both in vivo and in vitro infections, we have demonstrated that PEDV infection induces mitochondrial fission and leakage of mtDNA by upregulating Drp1 expression, and identified Smad3 as a crucial transcription factor responsible for regulating Drp1 expression. By establishing an inflammatory model using PEDV-infected cells, we have identified Drp1-mediated mtDNA release as an upstream event triggering TLR9/NF-κB pathway activation during PEDV infection. These findings provide novel insights into the relationship between PEDV infection and host inflammatory responses from the perspective of mitochondrial dynamics.
2026, 41(3): 602-611.
doi: 10.1016/j.virs.2026.05.008
Received: 13 March 2026
Accepted: 27 May 2026
Published: 29 May 2026
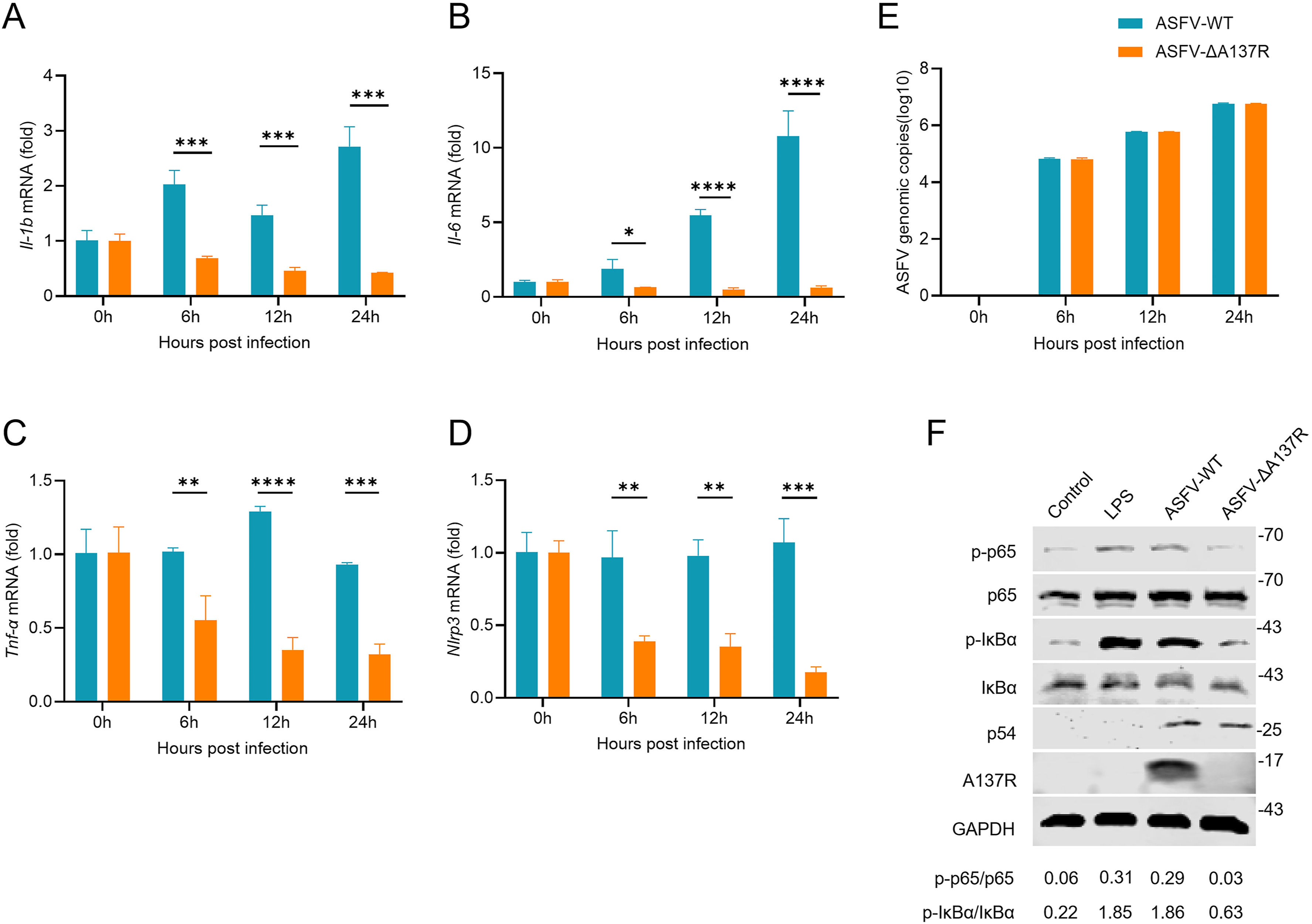
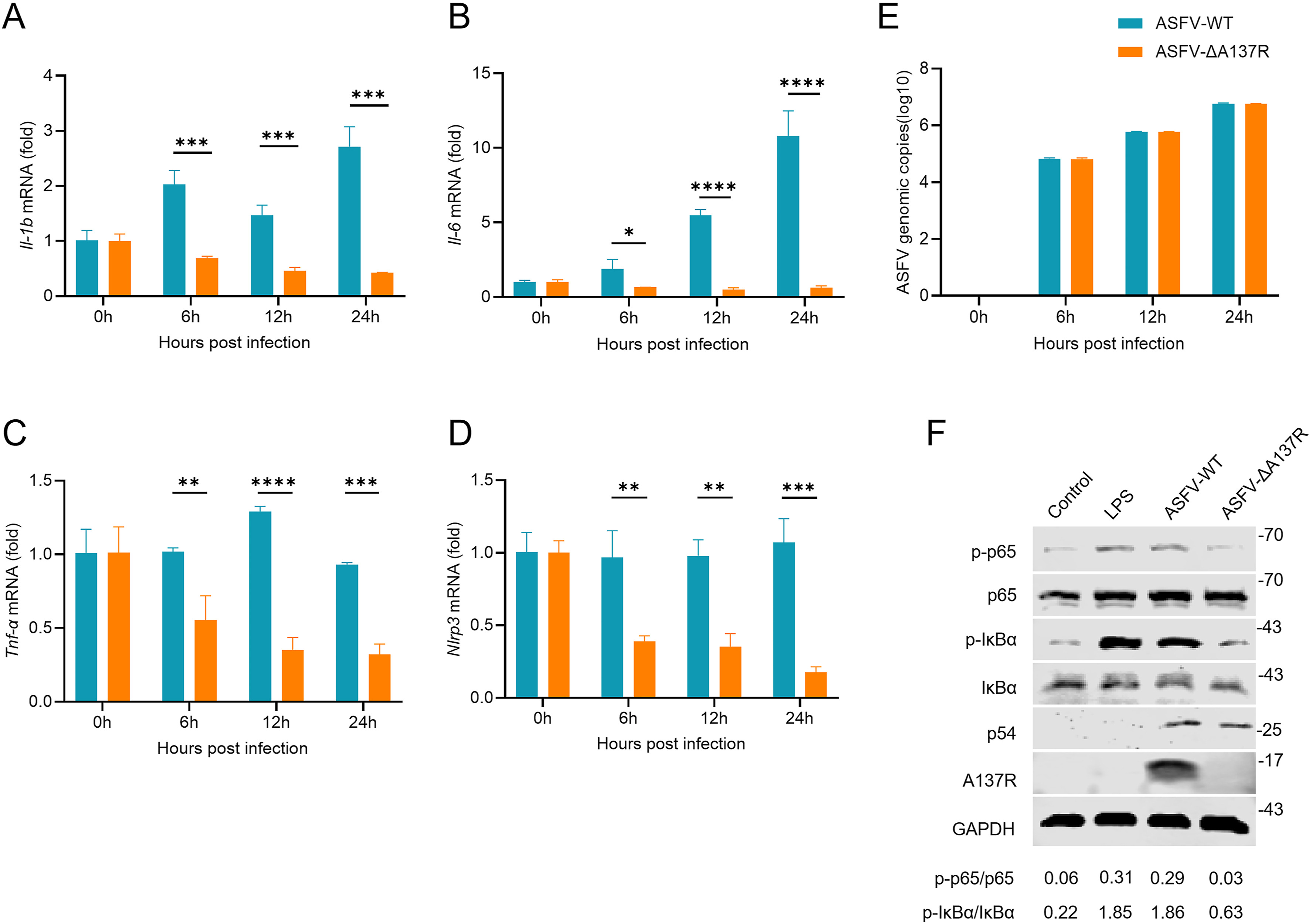
African swine fever (ASF), caused by the African swine fever virus (ASFV), is characterized by high mortality in infected pigs. ASFV infection triggers severe inflammatory response in the host, which is a crucial contributor to the high lethality of this disease. However, the underlying mechanism by which ASFV infection induces inflammatory response is still poorly understood. In this study, we found that UV-inactivated ASFV induces interleukin-1β (IL-1β) production, suggesting that certain structural proteins incorporated in the virion possess the ability to trigger inflammatory response. Further investigations demonstrated that deletion of the ASFV A137R gene significantly inhibited the ASFV-induced upregulation of the mRNA transcription of various proinflammatory genes and phosphorylation of p65 and IκBα. Furthermore, the purified pA137R protein promoted the mRNA transcription of these proinflammatory genes and phosphorylation of p65 and IκBα. Additionally, pA137R protein interacted with the NACHT and LRR domains of NLRP3 through its N terminal 1-99 amino acid domain, thereby promoting the oligomerization of NLRP3 and ASC and subsequently facilitating NLRP3 inflammasome assembly. Collectively, our findings identify ASFV pA137R protein as a key proinflammatory determinant of ASFV, which not only advances our understanding of the molecular mechanisms underlying ASFV-induced inflammatory response but also provides new insights into ASFV pathogenesis.
2026, 41(3): 612-623.
doi: 10.1016/j.virs.2026.06.005
Received: 05 December 2025
Accepted: 05 June 2026
Published: 09 June 2026
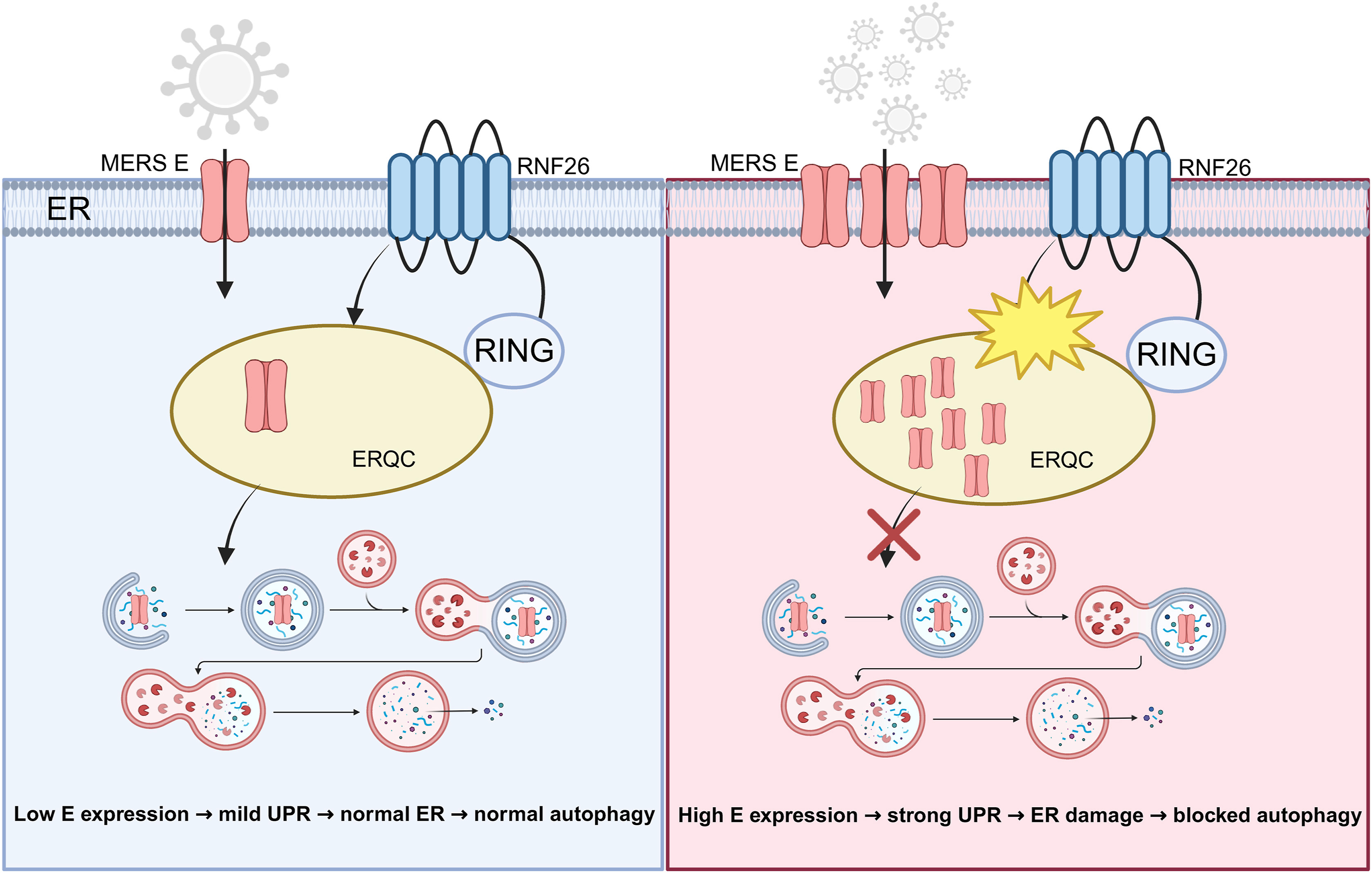
Coronavirus envelope (E) proteins are small, highly conserved viroporins essential for virion assembly and pathogenicity. Despite extensive characterization of their ion channel activity, how host cells sense and dispose of excessive viral membrane proteins remains poorly understood. Here we show that expression of the MERS-CoV E protein triggers pronounced ER stress and autophagy activation in human cells. The E protein is selectively degraded through an RNF26-dependent autophagy-lysosome pathway, and inhibition of autophagy or loss of RNF26 function leads to E accumulation and sustained unfolded protein response. Mechanistically, RNF26, an ER-anchored E3 ubiquitin ligase, promotes RING-dependent clearance of the viral protein through an ER protein quality control-associated pathway linked to autophagy and ER stress adaptation. Disruption of this process establishes a self-amplifying ER stress-autophagy feedback loop that exacerbates proteotoxicity. These findings define a membrane homeostatic conflict between viral viroporins and the host defense machinery, and identify RNF26 as a potential therapeutic target for mitigating viroporin-induced cytotoxicity through host-directed intervention.
2026, 41(3): 624-637.
doi: 10.1016/j.virs.2026.06.007
Received: 09 March 2026
Accepted: 10 June 2026
Published: 13 June 2026
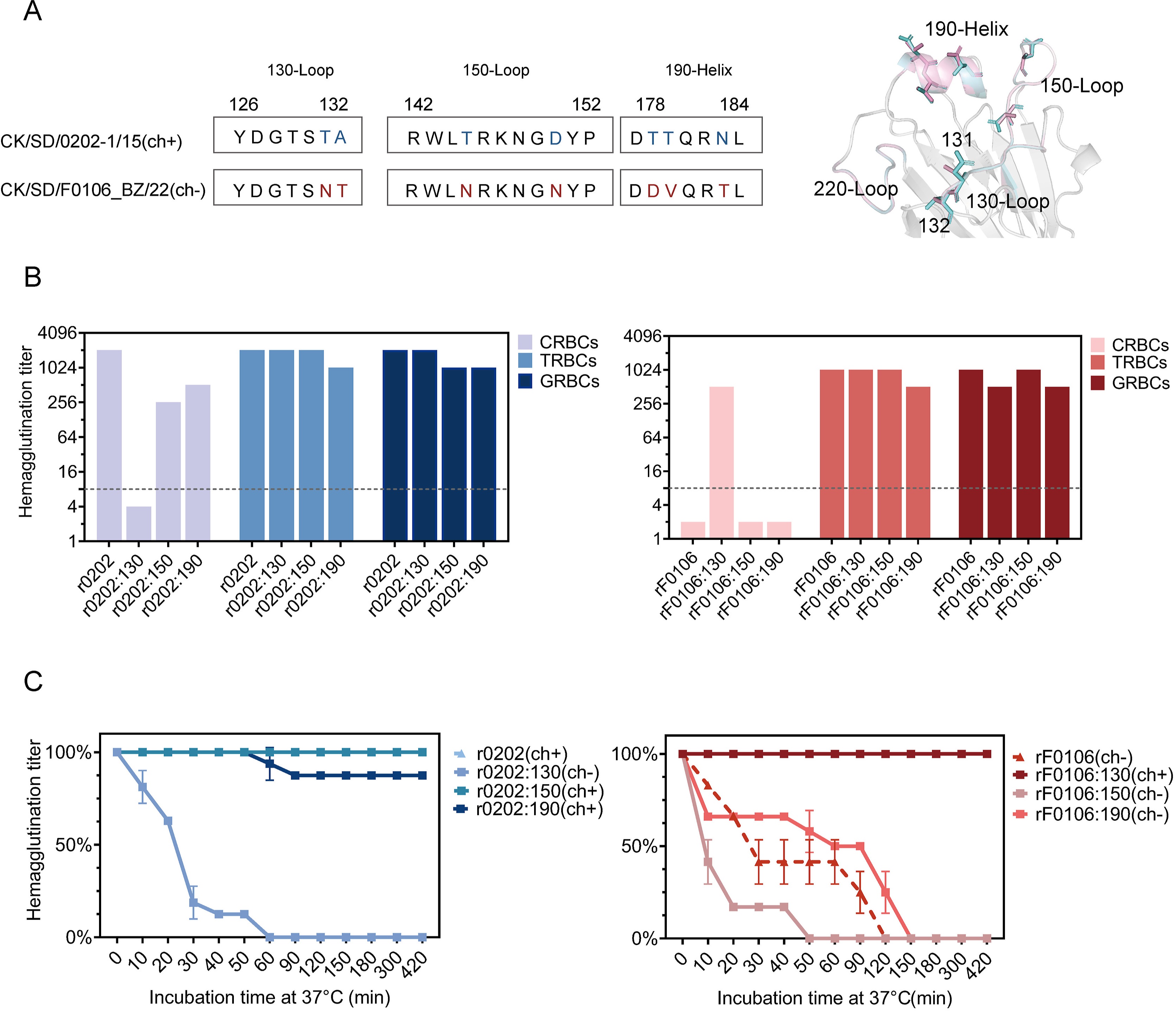
H9N2 avian influenza viruses (AIVs), which are enzootic in poultry and possess spillover potential to humans, represent a considerable global public health threat. Since 2021, we have observed an unprecedented decline in the ability of H9N2 viruses to agglutinate chicken red blood cells (CRBCs), a change that impairs effective detection in routine surveillance. Substituting CRBCs with turkey or guinea pig erythrocytes in hemagglutination assays effectively restored virus detectability. Through integrated bioinformatic and biological approaches, we identified that this phenotype is associated with specific amino acid substitutions at residues 131 and 132 of the 130-loop in the hemagglutinin (HA) protein. The HA motif 131/132-NT emerged in 2015 and rapidly increased in frequency, becoming the dominant pattern since 2021 in chickens and humans. Critically, the 131/132-NT motif attenuated viral binding affinity to short-chain α2,6-linked sialic acid receptors, explaining the loss of agglutination with CRBCs. Animal experiments further demonstrated that viruses carrying the HA 131/132-NT mutations maintained strong infectivity in chickens but altered tissue tropism in mice, showing reduced replication in the lungs. Collectively, our findings reveal a potential surveillance gap caused by reduced hemagglutination activity of H9N2 viruses, which may compromise the sensitivity of CRBC-based detection and potentially lead to under-detection of circulating viruses. Incorporating turkey or guinea pig red blood cells into surveillance protocols is therefore recommended to enhance detection sensitivity.
2026, 41(3): 638-650.
doi: 10.1016/j.virs.2026.06.008
Received: 12 April 2026
Accepted: 09 June 2026
Published: 13 June 2026
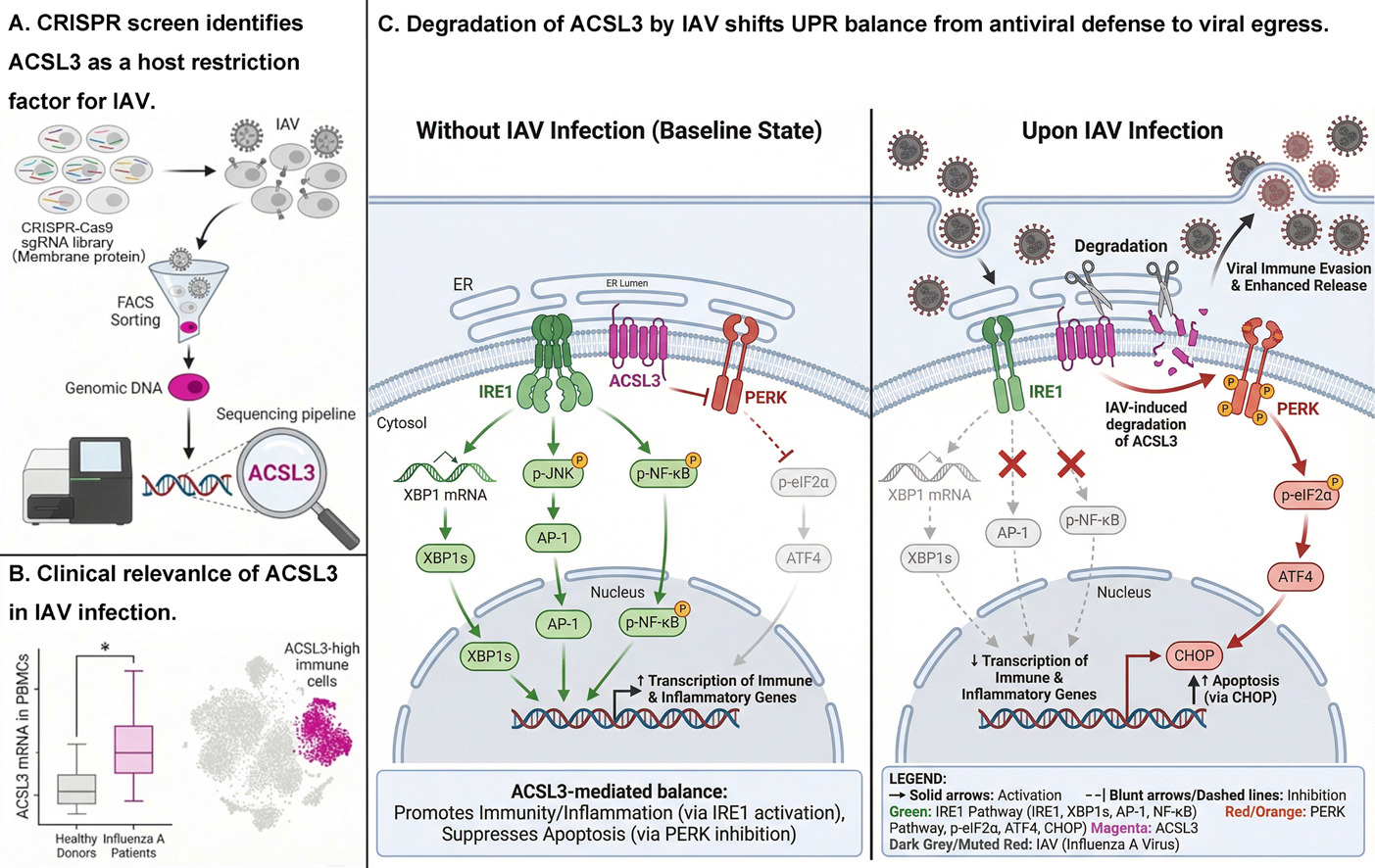
Host restriction factors and viral evasion strategies involved in the virus-host arms races remain to be discovered. Through an initial membrane protein-targeted CRISPR-Cas9 screening, we identified Acyl-CoA synthetase long-chain family member 3 (ACSL3) as a restriction factor against influenza A virus (IAV). Clinical transcriptomic analysis shows that ACSL3 is upregulated in peripheral blood mononuclear cells from mildly ill patients with influenza. Ectopic expression and loss-of-function validation demonstrate the physiological role of ACSL3 in restricting viral proliferation of diverse IAV subtypes (H1N1, H3N2, H3N8) in vitro and in vivo. Mechanistically, ACSL3 potentiates the antiviral unfolded protein response (UPR) by engaging the endoplasmic reticulum (ER) stress sensors IRE1α and PERK, thereby enhancing the IRE1α-XBP1-s driven inflammation and inhibiting the PERK-ATF4-CHOP mediated apoptosis. Conversely, IAV triggers the lysosomal degradation of ACSL3 and thus reprograms UPR to promote an immunologically silent apoptosis for viral egress and dissemination. Our findings establish ACSL3 as a molecular switch balancing the UPR-mediated antiviral response, and reveal a targeted viral evasion strategy in the virus-host interplay.
2026, 41(3): 651-661.
doi: 10.1016/j.virs.2026.05.006
Received: 03 February 2026
Accepted: 22 May 2026
Published: 29 May 2026
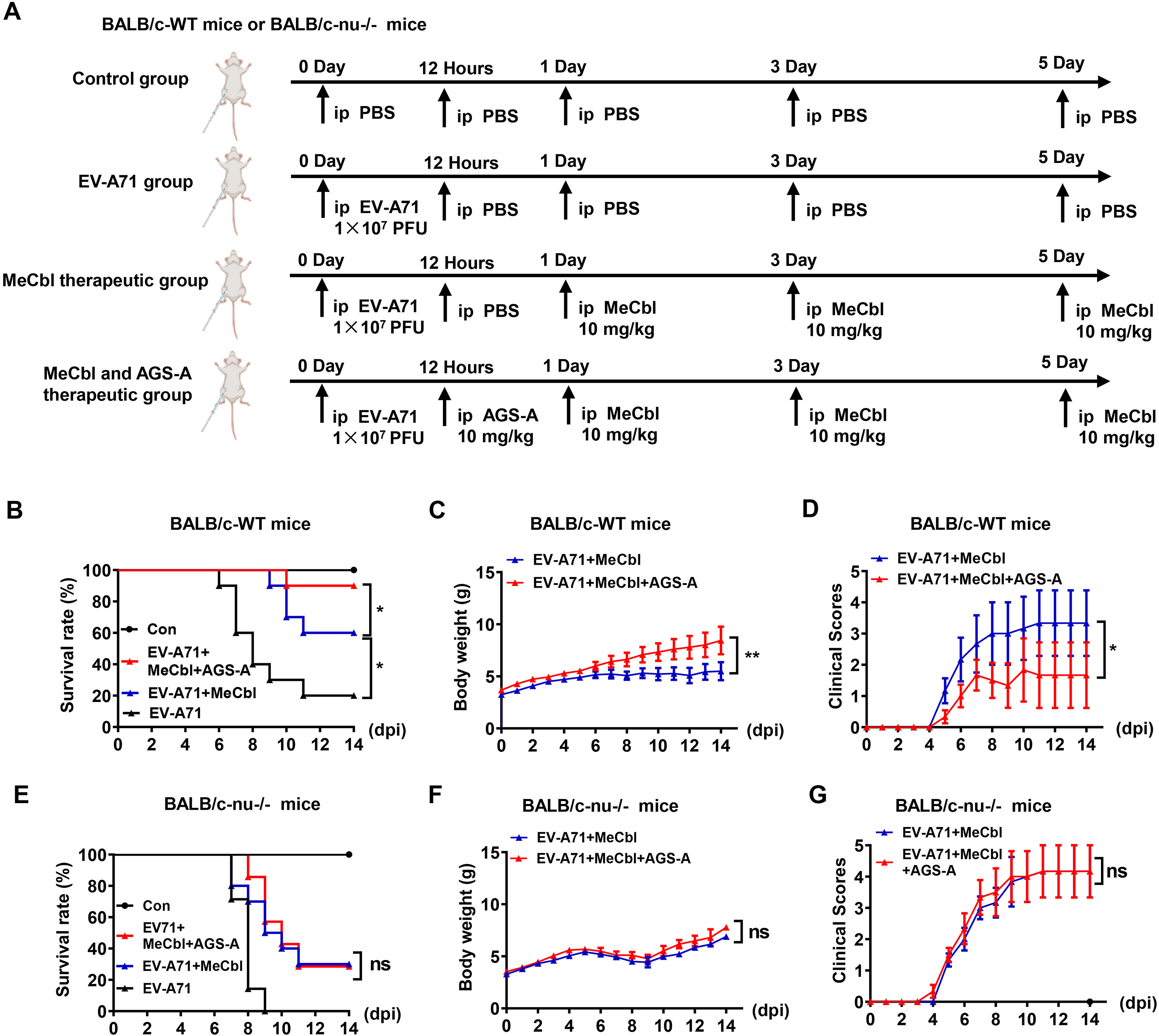
Enterovirus A71 (EV-A71) is the primary pathogen causing severe hand-foot-and-mouth disease (HFMD) in young children, with T-cell immune dysfunction closely linked to severe clinical outcomes. However, the molecular mechanisms underlying EV-A71-mediated T-cell impairment remain unclear, and no specific therapies are currently available. Here, we investigated the interaction between EV-A71 and T cells, and explored potential targeted therapeutic strategies. Our results showed that EV-A71 efficiently infects T cell lines (Jurkat, EL-4) and primary mouse CD3+ T cells in a dose- and time-dependent manner, inducing T-cell death and upregulating pro-inflammatory cytokines (IL-1β, IL-6, TNF-α). Mechanistically, EV-A71 infection triggers GSDME-dependent pyroptosis in T cells via caspase-3 activation, rather than GSDMD-dependent pyroptosis, as evidenced by genetic ablation and inhibitor experiments. Methylcobalamin (MeCbl), a specific GSDME inhibitor, rescued EV-A71-induced T-cell loss, and significantly improved the survival rate (80%) of EV-A71-infected newborn mice. Furthermore, the combined treatment with MeCbl and AGS-A (a T cell-dependent therapeutic agent) exerted a synergistic protective effect, achieving 90% survival rate in wild-type mice, which was abrogated in T cell-deficient BALB/c-nu-/- mice. Collectively, our findings identify GSDME-dependent T-cell pyroptosis as a key pathogenic mechanism of EV-A71 infection and highlight MeCbl as a promising targeted agent for HFMD treatment, either used alone or in combination with AGS-A.
2026, 41(3): 662-675.
doi: 10.1016/j.virs.2026.06.002
Received: 07 December 2025
Accepted: 01 June 2026
Published: 03 June 2026
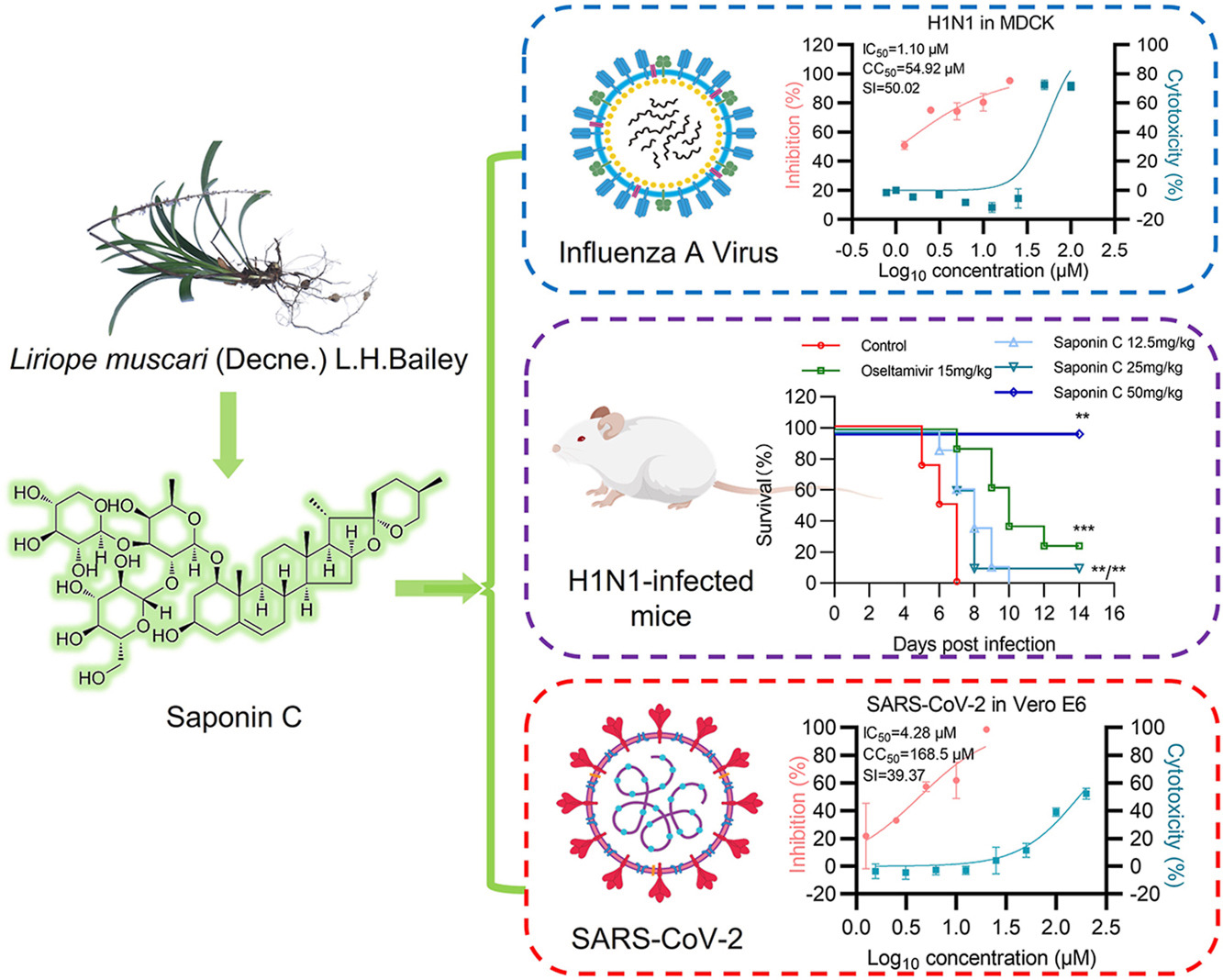
Saponin C (also known as DT-13) is the dominant ingredient extracted from the tuber of Liriope muscari (Decne.) L. H. Bailey, which exhibits multiple pharmacological activities, including anti-tumor, anti-thrombotic, cardioprotective and anti-inflammatory effects. However, its antiviral potential remains unexplored. This study aimed to investigate the inhibitory activity and underlying mechanisms of Saponin C against both influenza A virus (IAV) and SARS-CoV-2. In vitro experiments demonstrated that Saponin C dose-dependently inhibited IAV replication in MDCK and A549 cells. Time-of-addition assay revealed that Saponin C targeted the post-entry stages of IAV replication by impairing viral ribonucleoprotein (vRNP) function. This was evidenced by the suppression of viral polymerase activity, reduction of viral RNA synthesis, and delayed nuclear export of vRNP. The in vivo efficacy was further validated in a lethal H1N1 mouse model, where both prophylactic and therapeutic administration of Saponin C significantly improved the survival rate of infected mice, reduced viral loads in lung tissues, and attenuated virus-induced pulmonary inflammation in a dose-dependent manner. Beyond IAV, Saponin C also effectively inhibited SARS-CoV-2 infection by blocking viral entry, specifically through interfering with the binding of viral spike protein to the human ACE2 receptor and suppressing spike-mediated membrane fusion. Collectively, these findings demonstrate that Saponin C possesses antiviral potential against IAV and SARS-CoV-2, and may thus serve as a promising therapeutic agent for the treatment of respiratory viral infections.
2026, 41(3): 676-687.
doi: 10.1016/j.virs.2026.06.003
Received: 18 January 2026
Accepted: 01 June 2026
Published: 04 June 2026
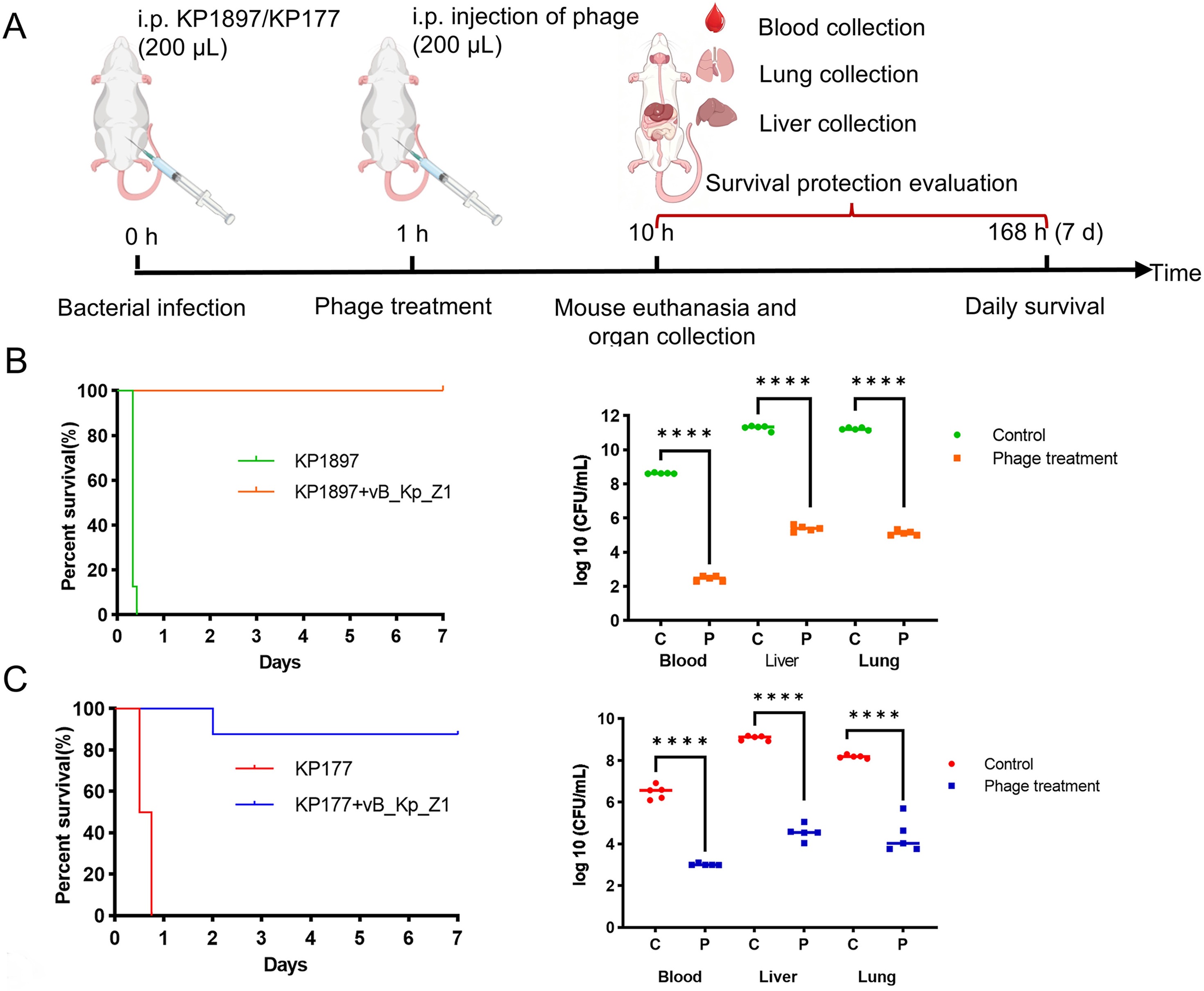
Klebsiella pneumoniae is an important opportunistic pathogen in both humans and animals. Controlling it has become increasingly difficult due to the rapid spread of antimicrobial resistance. In this study, we isolated and characterized a novel lytic bacteriophage, vB_Kp_Z1, and evaluated its therapeutic efficacy against K1-serotype K. pneumoniae. Host range analysis showed that vB_Kp_Z1 was strictly specific to K1 strains, as confirmed across multiple prevalent capsular types. The in vivo efficacy of vB_Kp_Z1 was assessed using intraperitoneal infection models in mice. Two hypervirulent K1 strains were used: a pigeon-derived strain (KP1897) and a human clinical strain (KP177). Phage treatment significantly improved survival compared with phosphate-buffered saline-treated controls. It provided complete protection in KP1897-infected mice and achieved an 87.5% survival rate in KP177-infected mice. In addition, phage administration markedly reduced bacterial loads in the blood, liver, and lungs, indicating effective control of systemic dissemination. These findings demonstrate that vB_Kp_Z1 is a K1-specific bacteriophage with therapeutic potential against hypervirulent K. pneumoniae, including strains from different host species.
A C-type single-domain antibody with protective efficacy against H1N1 via respiratory administration
2026, 41(3): 688-699.
doi: 10.1016/j.virs.2026.05.007
Received: 03 March 2026
Accepted: 25 May 2026
Published: 28 May 2026
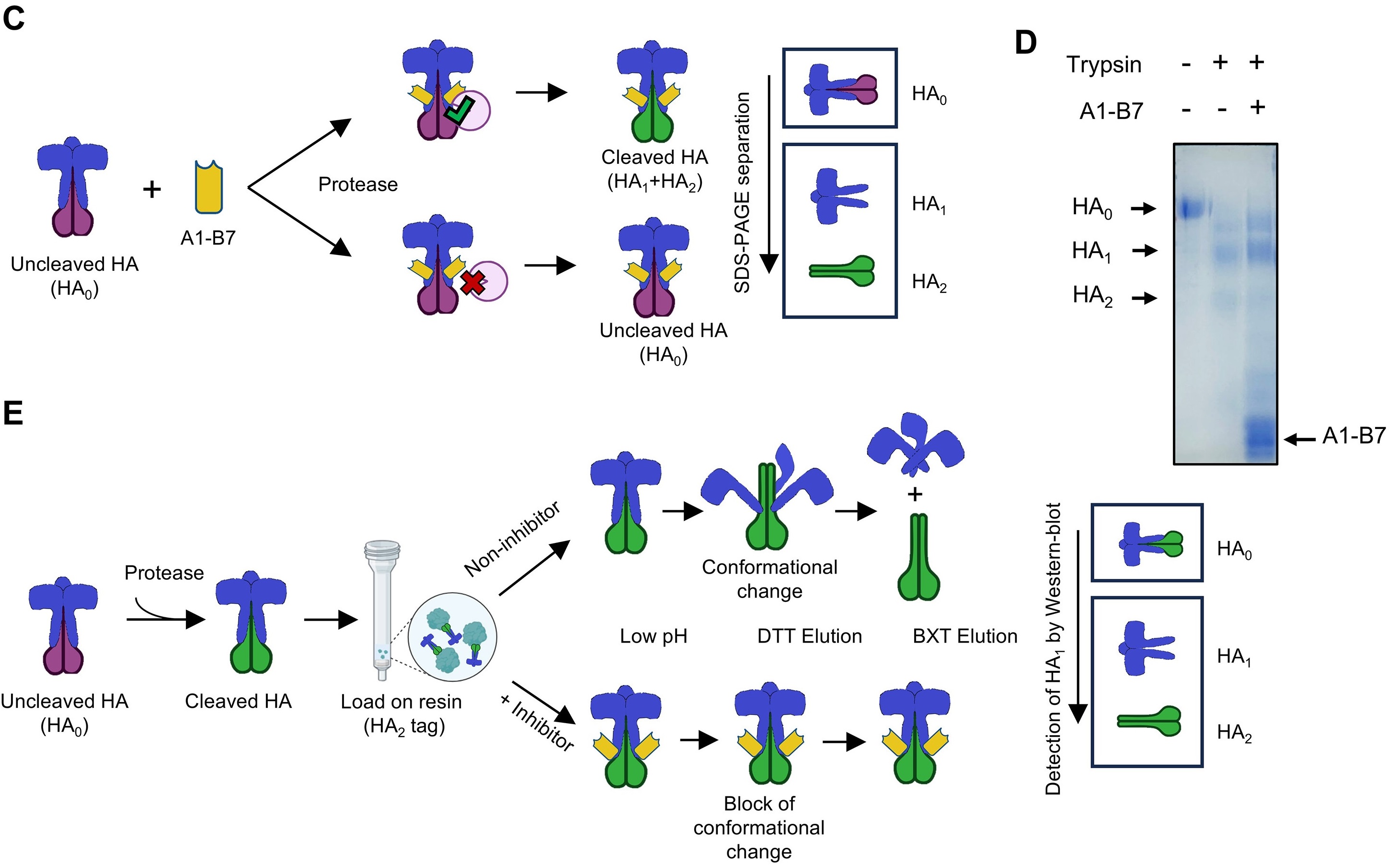
Influenza A viruses, particularly H1N1, pose a significant public health threat, highlighting the continued need for novel prophylactics and therapeutics. Monoclonal antibodies are promising, but conventional IgGs face challenges in respiratory delivery due to their large size. This study presents A1-B7, a C-type single-domain antibody (C-sdAb) based on a scaffold derived from a human IgG1 CH2 domain, targeting the hemagglutinin (HA) of H1N1 influenza virus. A1-B7 potently neutralizes multiple H1N1 subtypes in vitro. Both prophylactic intranasal administration and therapeutic aerosol inhalation of A1-B7 confer complete protection and reduce live virus in the lungs to undetectable levels. Structural prediction and epitope mapping reveal that A1-B7 binds a conserved stem-region epitope, engaging the HA2 A-helix and a hydrophobic groove. A1-B7 can block low-pH induced conformational change of HA. These findings establish A1-B7 as a promising candidate for further development as a convenient, respiratory tract delivered countermeasure against influenza.
2026, 41(3): 700-710.
doi: 10.1016/j.virs.2026.06.011
Received: 10 December 2025
Accepted: 16 June 2026
Published: 19 June 2026
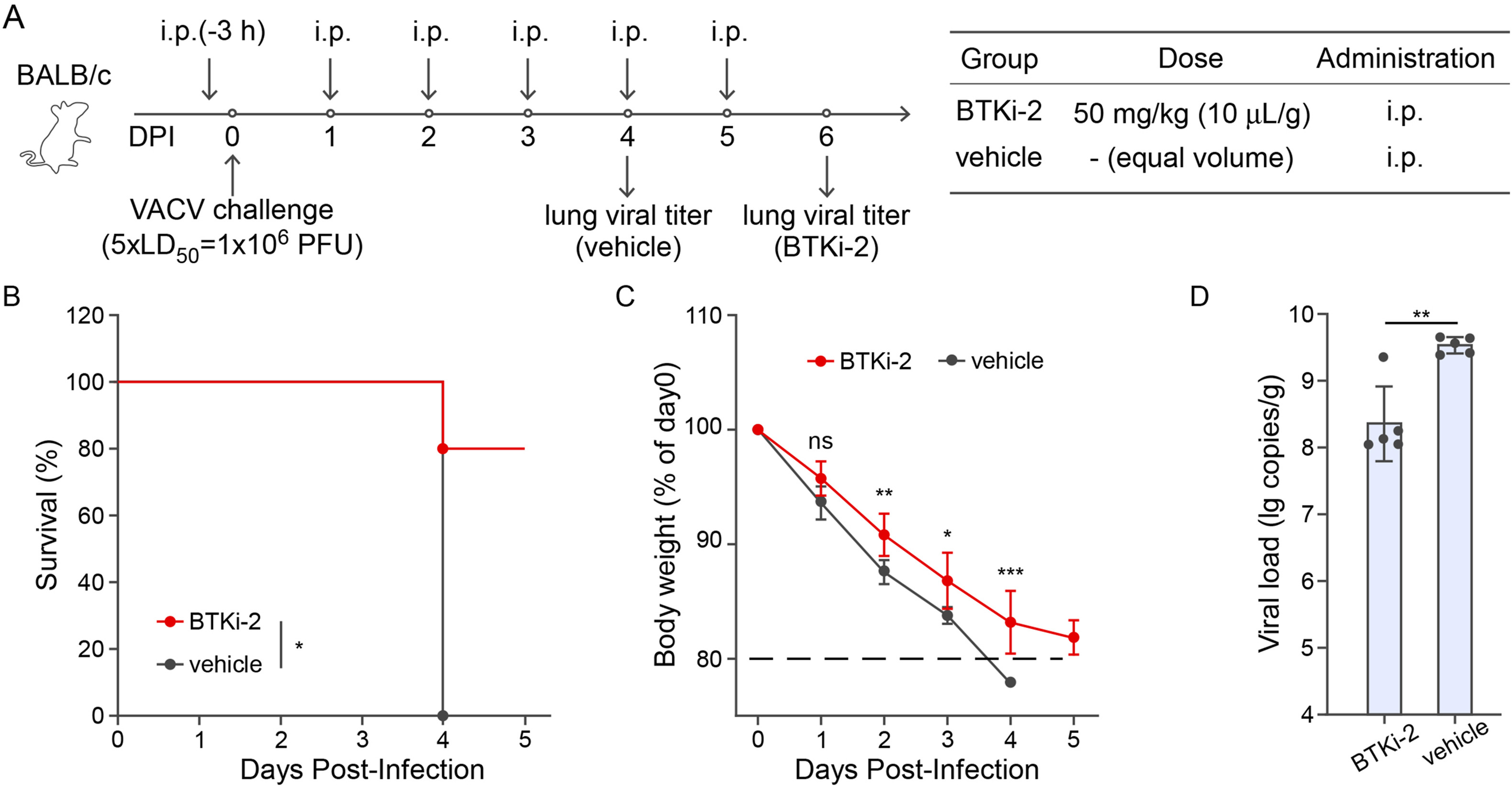
The recent global outbreak of mpox virus (MPXV) infections underscores the urgent need for antiviral therapies against orthopoxviruses. In this study, using a high-content screening (HCS) platform based on a modified vaccinia virus Tiantan strain with GFP insertion (MVTT-GFP) under BSL-2 conditions, we screened 1513 kinase inhibitors for antiviral activity. Among these, Bruton's tyrosine kinase inhibitor BTKi-2 emerged as a potent candidate, exhibiting IC50 of 0.535 μM against vaccinia virus (VACV) and 0.260 μM against MPXV in vitro, while maintaining low cytotoxicity. In a murine model of VACV-induced pneumonia, BTKi-2 treatment reduced lung viral loads by 90% and a significantly improved survival compared to vehicle-treated controls. Notably, mechanistic studies indicate that BTKi-2's antiviral effects cannot be completely attributed to the inhibition of BTK or EGFR/ErbB2 signaling. These findings highlight BTKi-2 as a promising antiviral agent in vitro and in vivo, suggesting that BTKi-2 may offer a potential avenue for future therapeutic development against orthopoxvirus infections.
2026, 41(3): 711-715.
doi: 10.1016/j.virs.2026.05.005
Received: 09 February 2026
Accepted: 21 May 2026
Published: 27 May 2026
Highlights
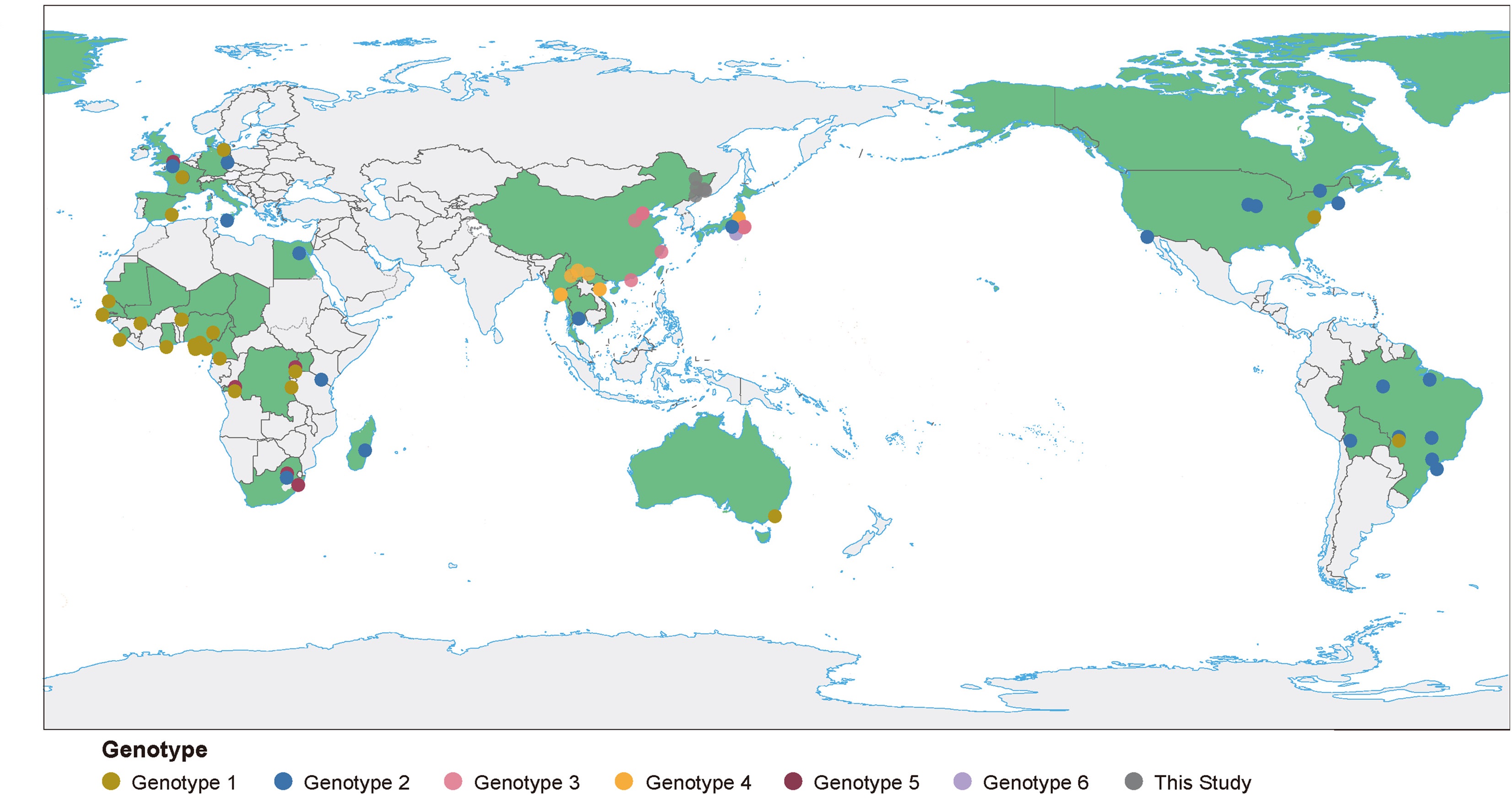
1 First report of HPgV genotype 3 strains in Heilongjiang, China, revealing its genetic diversity in East Asia.
2 Phylogeographic analysis demonstrated spatial clustering in the distribution of human pegivirus genotypes 1-6.
3 Identified an anomalous evolutionary branch in the NS4A region and ten specific amino acid mutation sites.
4 Subgenomic alignments yield different lineage calls, necessitating full-length genomes for accurate genotyping.
1 First report of HPgV genotype 3 strains in Heilongjiang, China, revealing its genetic diversity in East Asia.
2 Phylogeographic analysis demonstrated spatial clustering in the distribution of human pegivirus genotypes 1-6.
3 Identified an anomalous evolutionary branch in the NS4A region and ten specific amino acid mutation sites.
4 Subgenomic alignments yield different lineage calls, necessitating full-length genomes for accurate genotyping.
2026, 41(3): 716-719.
doi: 10.1016/j.virs.2026.05.010
Received: 14 March 2026
Accepted: 29 May 2026
Published: 03 June 2026
Highlights
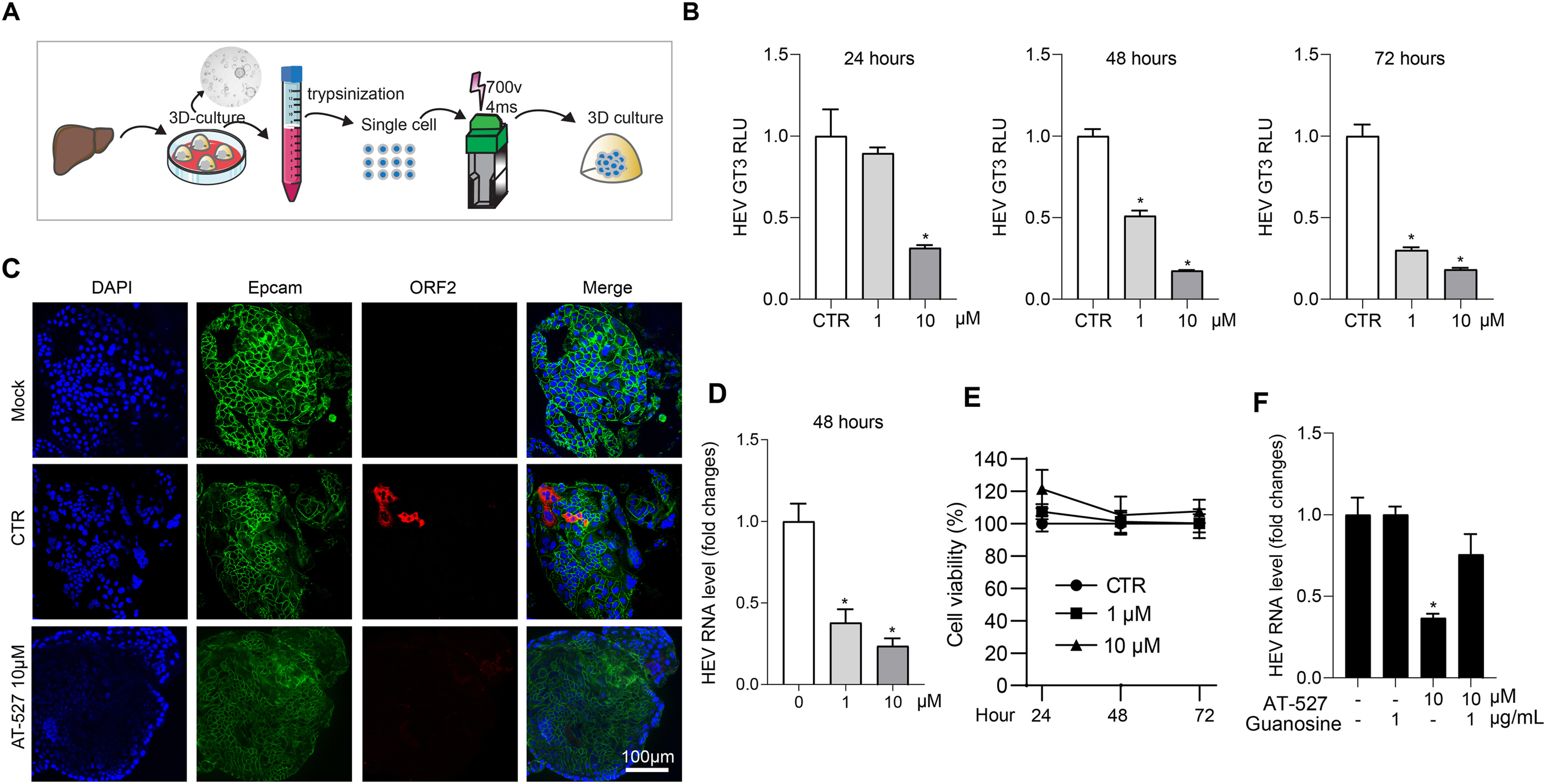
1 The nucleotide analog bemnifosbuvir (AT-527) inhibits hepatitis E virus (HEV) infection in cell and organoid models.
2 Supplementation with guanosine largely reversed the anti-HEV activity of AT-527.
3 Combining AT-527 with ribavirin or interferon-α resulted in synergistic antiviral effects.
1 The nucleotide analog bemnifosbuvir (AT-527) inhibits hepatitis E virus (HEV) infection in cell and organoid models.
2 Supplementation with guanosine largely reversed the anti-HEV activity of AT-527.
3 Combining AT-527 with ribavirin or interferon-α resulted in synergistic antiviral effects.
2026, 41(3): 720-723.
doi: 10.1016/j.virs.2026.06.009
Received: 03 April 2026
Accepted: 08 June 2026
Published: 13 June 2026
Highlights
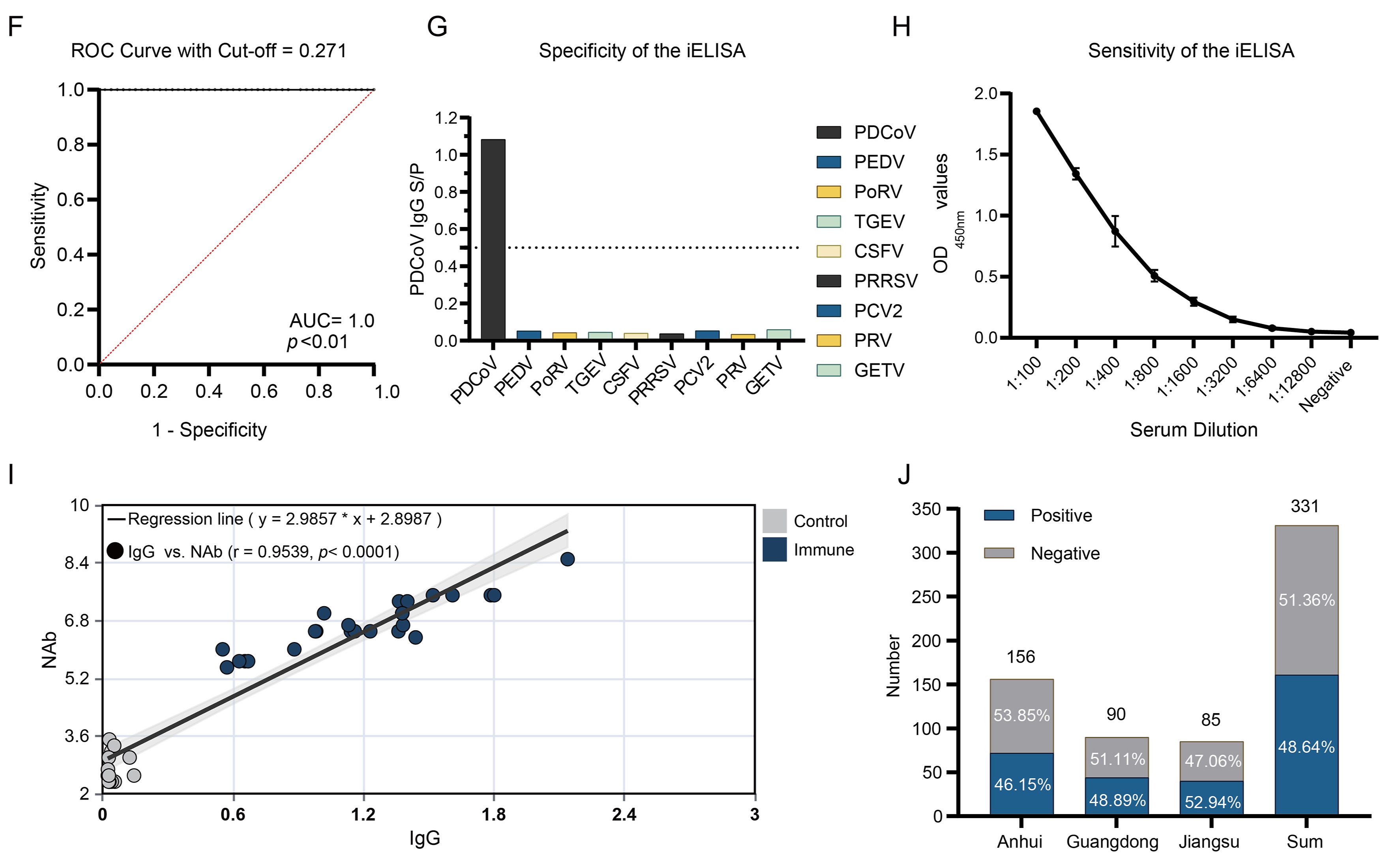
1 A novel iELISA utilizing ferritin-nanoparticle-displayed PDCoV S protein was established.
2 Excellent diagnostic performance with high sensitivity, specificity, and correlation with NAb titers.
3 Revealed widespread PDCoV circulation (48.64%) in the surveyed provinces of China, supporting field epidemiological studies.
1 A novel iELISA utilizing ferritin-nanoparticle-displayed PDCoV S protein was established.
2 Excellent diagnostic performance with high sensitivity, specificity, and correlation with NAb titers.
3 Revealed widespread PDCoV circulation (48.64%) in the surveyed provinces of China, supporting field epidemiological studies.
2026, 41(3): 724-727.
doi: 10.1016/j.virs.2026.06.012
Received: 14 May 2026
Accepted: 17 June 2026
Published: 19 June 2026
Highlights
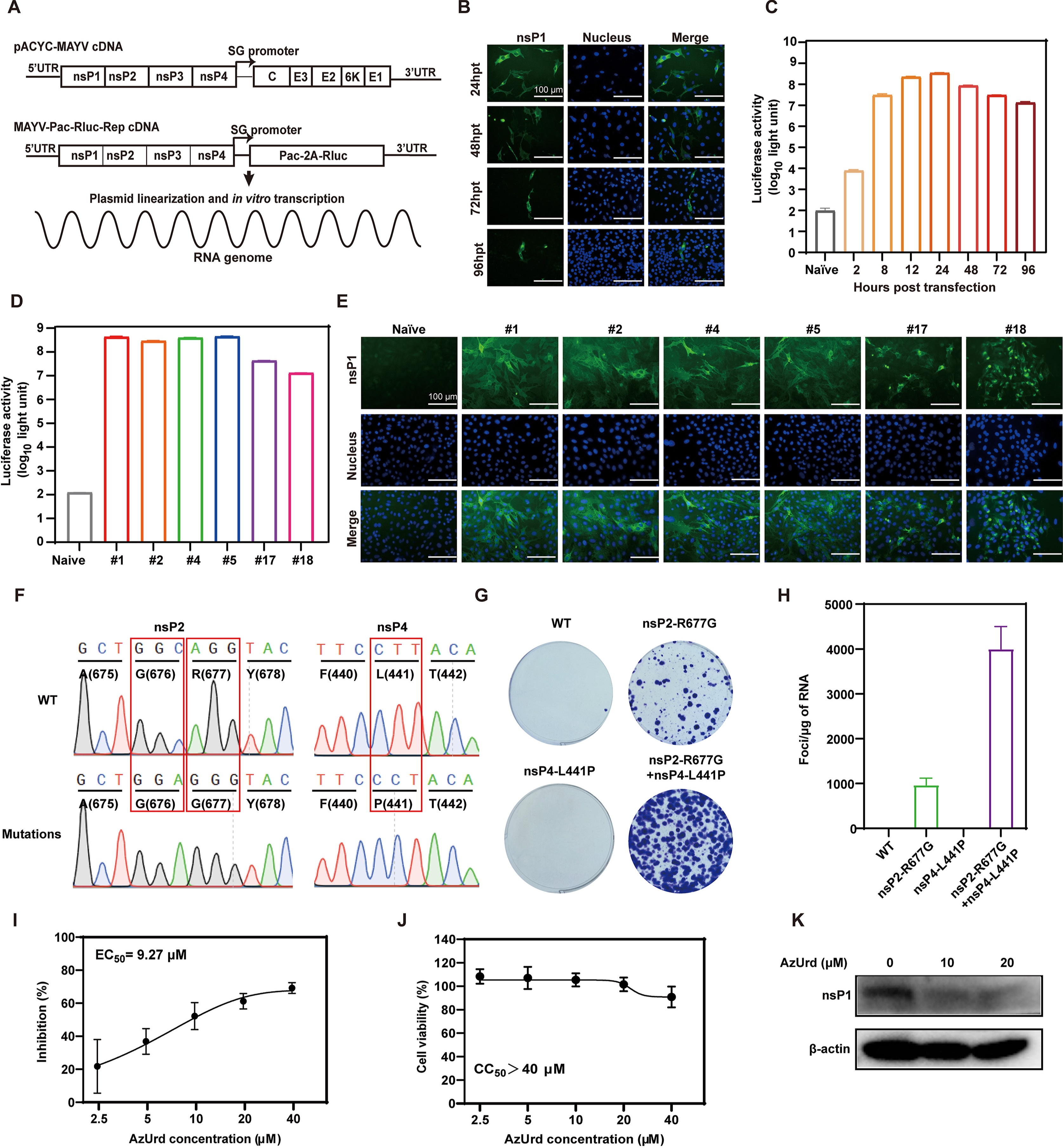
1 Construction of a MAYV replicon expressing both PAC and Rluc genes.
2 A stable BHK-21 cell line containing MAYV-Rep with puromycin selection was obtained.
3 A robust replicon cell-based HTS assay based was established.
1 Construction of a MAYV replicon expressing both PAC and Rluc genes.
2 A stable BHK-21 cell line containing MAYV-Rep with puromycin selection was obtained.
3 A robust replicon cell-based HTS assay based was established.
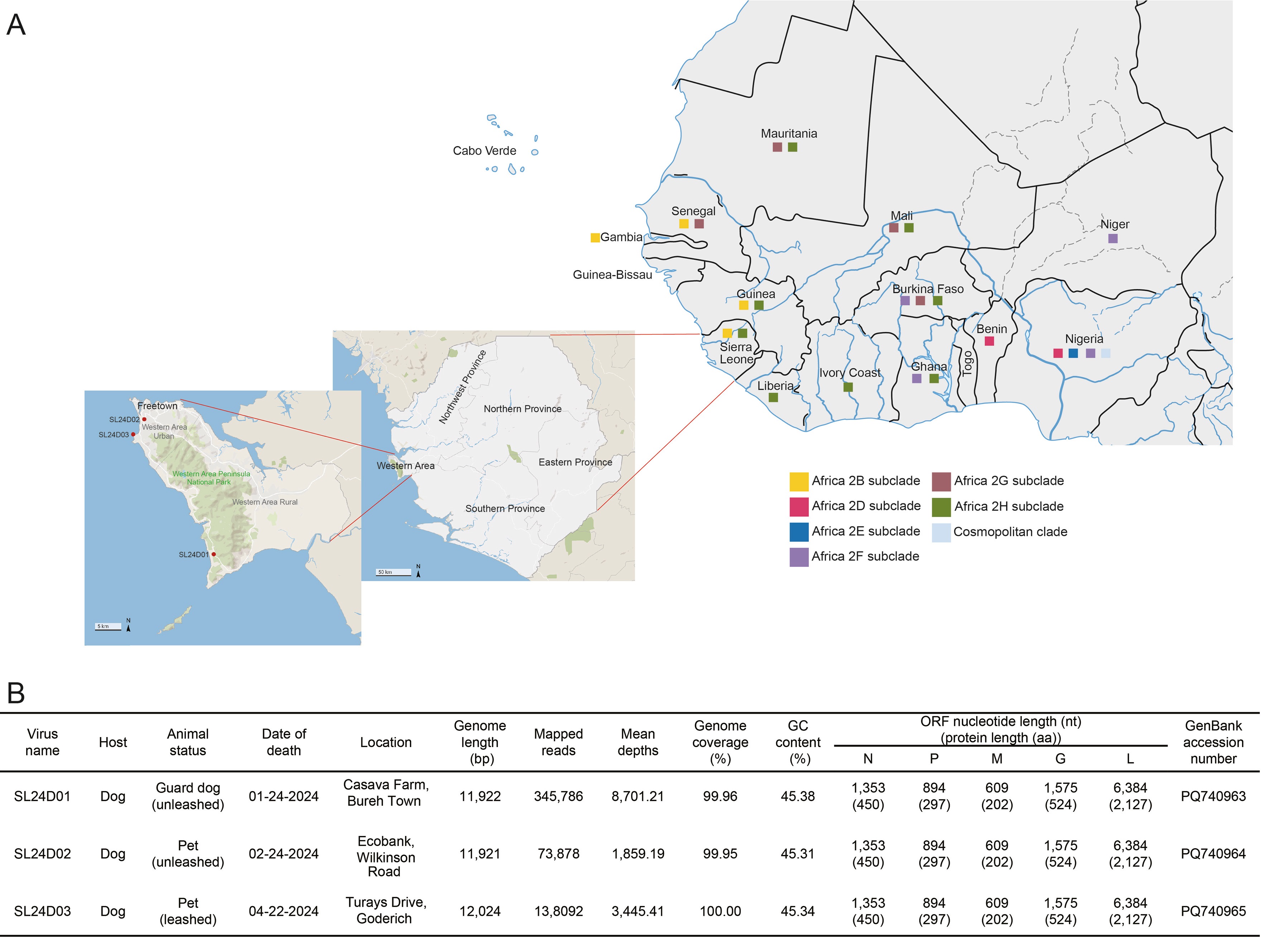
2026, 41(3): 728-731.
doi: 10.1016/j.virs.2026.06.010
Received: 05 June 2025
Accepted: 15 June 2026
Published: 17 June 2026
Highlights
1 First set of three complete genomic sequences of RABV strains was obtained from Sierra Leone.
2 Phylogenetic analysis identified the endemic Africa 2H subclade with high homology to those from neighboring countries.
3 Low dog vaccination coverage underscores urgent need for coordinated vaccination and surveillance.
1 First set of three complete genomic sequences of RABV strains was obtained from Sierra Leone.
2 Phylogenetic analysis identified the endemic Africa 2H subclade with high homology to those from neighboring countries.
3 Low dog vaccination coverage underscores urgent need for coordinated vaccination and surveillance.