RNA barcode segments for SARS-CoV-2 identification from HCoVs and SARSr-CoV-2 lineages
Severe acute respiratory syndrome coronavirus 2 (SARS-CoV-2), the pathogen responsible for coronavirus disease 2019 (COVID-19), continues to evolve, giving rise to more variants and global reinfections. Previous research has demonstrated that barcode segments can effectively and cost-efficiently identify specific species within closely related populations. In this study, we designed and tested RNA barcode segments based on genetic evolutionary relationships to facilitate the efficient and accurate identification of SARS-CoV-2 from extensive virus samples, including human coronaviruses (HCoVs) and SARSr-CoV-2 lineages. Nucleotide sequences sourced from NCBI and GISAID were meticulously selected and curated to construct training sets, encompassing 1733 complete genome sequences of HCoVs and SARSr-CoV-2 lineages. Through genetic-level species testing, we validated the accuracy and reliability of the barcode segments for identifying SARS-CoV-2. Subsequently, 75 main and subordinate species-specific barcode segments for SARS-CoV-2, located in ORF1ab, S, E, ORF7a, and N coding sequences, were intercepted and screened based on single-nucleotide polymorphism sites and weighted scores. Post-testing, these segments exhibited high recall rates (nearly 100%), specificity (almost 30% at the nucleotide level), and precision (100%) performance on identification. They were eventually visualized using one and two-dimensional combined barcodes and deposited in an online database (http://virusbarcodedatabase.top/). The successful integration of barcoding technology in SARS-CoV-2 identification provides valuable insights for future studies involving complete genome sequence polymorphism analysis. Moreover, this cost-effective and efficient identification approach also provides valuable reference for future research endeavors related to virus surveillance.
more >>
Interconnection of cellular autophagy and endosomal vesicle trafficking and its role in hepatitis B virus replication and release
Hepatitis B virus (HBV) produces and releases various particle types, including complete virions, subviral particles with envelope proteins, and naked capsids. Recent studies demonstrate that HBV exploits distinct intracellular membrane trafficking pathways, including the endosomal vesicle trafficking and autophagy pathway, to assemble and release viral and subviral particles. Herein, we summarize the findings about the distinct roles of autophagy and endosomal membrane trafficking and the interaction of both pathways in HBV replication, assembly, and release.
more >>
Classifying hepatitis B therapies with insights from covalently closed circular DNA dynamics
The achievement of a functional cure for chronic hepatitis B (CHB) remains limited to a minority of patients treated with currently approved drugs. The primary objective in developing new anti-HBV drugs is to enhance the functional cure rates for CHB. A critical prerequisite for the functional cure of CHB is a substantial reduction, or even eradication of covalently closed circular DNA (cccDNA). Within this context, the changes in cccDNA levels during treatment become as a pivotal concern. We have previously analyzed the factors influencing cccDNA dynamics and introduced a preliminary classification of hepatitis B treatment strategies based on these dynamics. In this review, we employ a systems thinking perspective to elucidate the fundamental aspects of the HBV replication cycle and to rationalize the classification of treatment strategies according to their impact on the dynamic equilibrium of cccDNA. Building upon this foundation, we categorize current anti-HBV strategies into two distinct groups and advocate for their combined use to significantly reduce cccDNA levels within a well-defined timeframe.
more >>
Ferroptosis contributes to JEV-induced neuronal damage and neuroinflammation
Ferroptosis is a newly discovered prototype of programmed cell death (PCD) driven by iron-dependent phospholipid peroxidation accumulation, and it has been linked to numerous organ injuries and degenerative pathologies. Although studies have shown that a variety of cell death processes contribute to JEV-induced neuroinflammation and neuronal injury, there is currently limited research on the specific involvement of ferroptosis. In this study, we explored the neuronal ferroptosis induced by JEV infection in vitro and in vivo. Our results indicated that JEV infection induces neuronal ferroptosis through inhibiting the function of the antioxidant system mediated by glutathione (GSH)/glutathione peroxidase 4 (GPX4), as well as by promoting lipid peroxidation mediated by yes-associated protein 1 (YAP1)/long-chain acyl-CoA synthetase 4 (ACSL4). Further analyses revealed that JEV E and prM proteins function as agonists, inducing ferroptosis. Moreover, we found that treatment with a ferroptosis inhibitor in JEV-infected mice reduces the viral titers and inflammation in the mouse brains, ultimately improving the survival rate of infected mice. In conclusion, our study unveils a critical role of ferroptosis in the pathogenesis of JEV, providing new ideas for the prevention and treatment of viral encephalitis.
more >>
Phosphorylation of PB2 at serine 181 restricts viral replication and virulence of the highly pathogenic H5N1 avian influenza virus in mice
Influenza A virus (IAV) continues to pose a pandemic threat to public health, resulting a high mortality rate annually and during pandemic years. Posttranslational modification of viral protein plays a substantial role in regulating IAV infection. Here, based on immunoprecipitation (IP)-based mass spectrometry (MS) and purified virus-coupled MS, a total of 89 phosphorylation sites distributed among 10 encoded viral proteins of IAV were identified, including 60 novel phosphorylation sites. Additionally, for the first time, we provide evidence that PB2 can also be acetylated at site K187. Notably, the PB2 S181 phosphorylation site was consistently identified in both IP-based MS and purified virus-based MS. Both S181 and K187 are exposed on the surface of the PB2 protein and are highly conserved in various IAV strains, suggesting their fundamental importance in the IAV life cycle. Bioinformatic analysis results demonstrated that S181E/A and K187Q/R mimic mutations do not significantly alter the PB2 protein structure. While continuous phosphorylation mimicked by the PB2 S181E mutation substantially decreases viral fitness in mice, PB2 K187Q mimetic acetylation slightly enhances viral virulence in mice. Mechanistically, PB2 S181E substantially impairs viral polymerase activity and viral replication, remarkably dampens protein stability and nuclear accumulation of PB2, and significantly weakens IAV-induced inflammatory responses. Therefore, our study further enriches the database of phosphorylation and acetylation sites of influenza viral proteins, laying a foundation for subsequent mechanistic studies. Meanwhile, the unraveled antiviral effect of PB2 S181E mimetic phosphorylation may provide a new target for the subsequent study of antiviral drugs.
more >>
Rapid identification of full-length genome and tracing variations of monkeypox virus in clinical specimens based on mNGS and amplicon sequencing
The monkeypox virus (MPXV) has triggered a current outbreak globally. Genome sequencing of MPXV and rapid tracing of genetic variants will benefit disease diagnosis and control. It is a significant challenge but necessary to optimize the strategy and application of rapid full-length genome identification and to track variations of MPXV in clinical specimens with low viral loads, as it is one of the DNA viruses with the largest genome and the most AT-biased, and has a significant number of tandem repeats. Here we evaluated the performance of metagenomic and amplicon sequencing techniques, and three sequencing platforms in MPXV genome sequencing based on multiple clinical specimens of five mpox cases in Chinese mainland. We rapidly identified the full-length genome of MPXV with the assembly of accurate tandem repeats in multiple clinical specimens. Amplicon sequencing enables cost-effective and rapid sequencing of clinical specimens to obtain high-quality MPXV genomes. Third-generation sequencing facilitates the assembly of the terminal tandem repeat regions in the monkeypox virus genome and corrects a common misassembly in published sequences. Besides, several intra-host single nucleotide variations were identified in the first imported mpox case. This study offers an evaluation of various strategies aimed at identifying the complete genome of MPXV in clinical specimens. The findings of this study will significantly enhance the surveillance of MPXV.
more >>
HIF-1α promotes virus replication and cytokine storm in H1N1 virus-induced severe pneumonia through cellular metabolic reprogramming
The mortality of patients with severe pneumonia caused by H1N1 infection is closely related to viral replication and cytokine storm. However, the specific mechanisms triggering virus replication and cytokine storm are still not fully elucidated. Here, we identified hypoxia inducible factor-1α (HIF-1α) as one of the major host molecules that facilitates H1N1 virus replication followed by cytokine storm in alveolar epithelial cells. Specifically, HIF-1α protein expression is upregulated after H1N1 infection. Deficiency of HIF-1α attenuates pulmonary injury, viral replication and cytokine storm in vivo. In addition, viral replication and cytokine storm were inhibited after HIF-1α knockdown in vitro. Mechanistically, the invasion of H1N1 virus into alveolar epithelial cells leads to a shift in glucose metabolism to glycolysis, with rapid production of ATP and lactate. Inhibition of glycolysis significantly suppresses viral replication and inflammatory responses. Further analysis revealed that H1N1-induced HIF-1α can promote the expression of hexokinase 2 (HK2), the key enzyme of glycolysis, and then not only provide energy for the rapid replication of H1N1 virus but also produce lactate, which reduces the accumulation of the MAVS/RIG-I complex and inhibits IFN-α/β production. In conclusion, this study demonstrated that the upregulation of HIF-1α by H1N1 infection augments viral replication and cytokine storm by cellular metabolic reprogramming toward glycolysis mainly through upregulation of HK2, providing a theoretical basis for finding potential targets for the treatment of severe pneumonia caused by H1N1 infection.
more >>
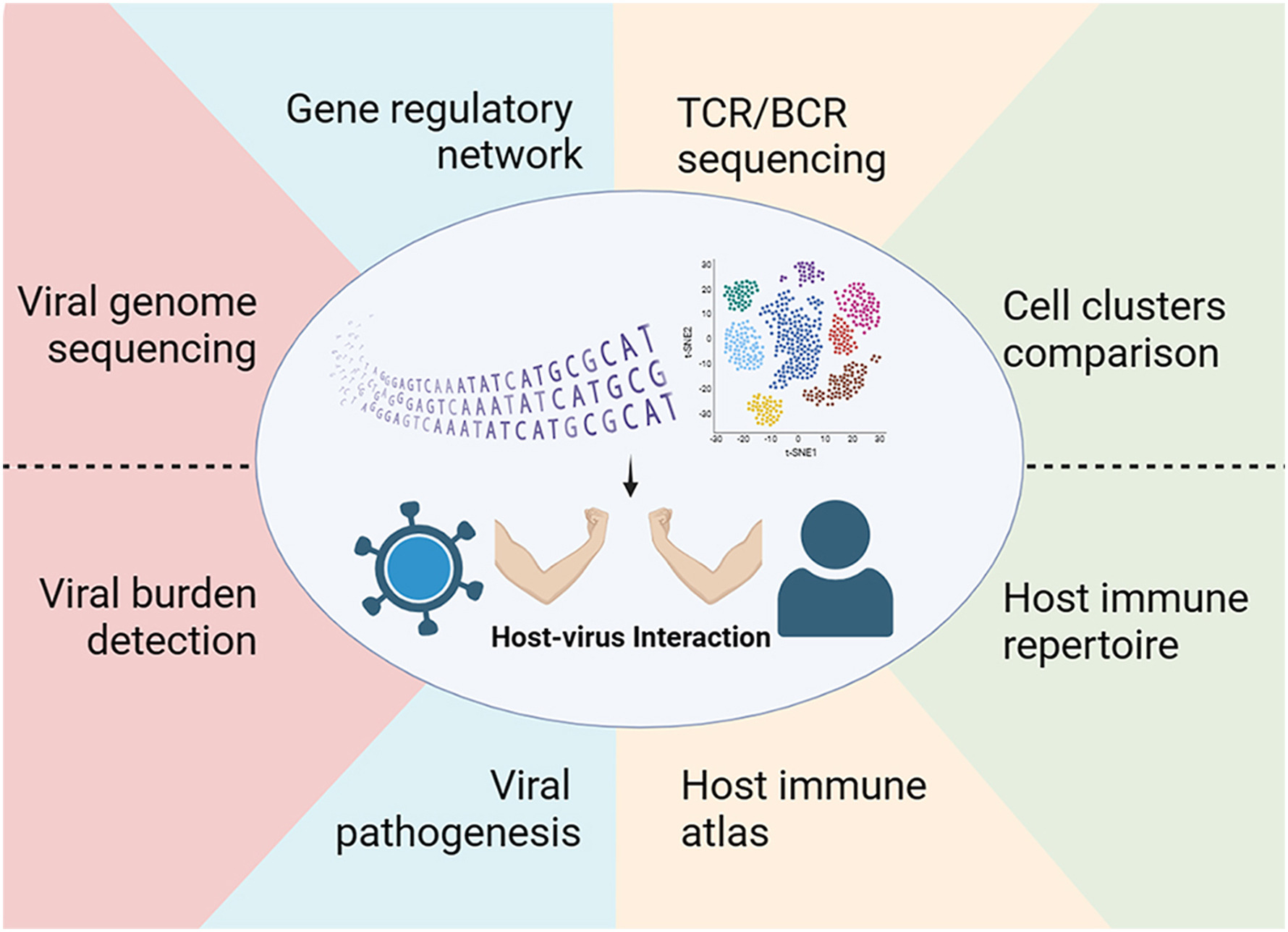
Single-cell RNA sequencing to understand host-virus interactions
Single-cell RNA sequencing (scRNA-seq) has allowed for the profiling of host and virus transcripts and host-virus interactions at single-cell resolution. This review summarizes the existing scRNA-seq technologies together with their strengths and weaknesses. The applications of scRNA-seq in various virological studies are discussed in depth, which broaden the understanding of the immune atlas, host-virus interactions, and immune repertoire. scRNA-seq can be widely used for virology in the near future to better understand the pathogenic mechanisms and discover more effective therapeutic strategies.
more >>
Hsp90 β is critical for the infection of severe fever with thrombocytopenia syndrome virus
Severe fever with thrombocytopenia syndrome (SFTS) caused by the SFTS virus (SFTSV) is an emerging disease in East Asia with a fatality rate of up to 30%. However, the viral-host interaction of SFTSV remains largely unknown. The heat-shock protein 90 (Hsp90) family consists of highly conserved chaperones that fold and remodel proteins and has a broad impact on the infection of many viruses. Here, we showed that Hsp90 is an important host factor involved in SFTSV infection. Hsp90 inhibitors significantly reduced SFTSV replication, viral protein expression, and the formation of inclusion bodies consisting of nonstructural proteins (NSs). Among viral proteins, NSs appeared to be the most reduced when Hsp90 inhibitors were used, and further analysis showed that their translation was affected. Co-immunoprecipitation of NSs with four isomers of Hsp90 showed that Hsp90 β specifically interacted with them. Knockdown of Hsp90 β expression also inhibited replication of SFTSV. These results suggest that Hsp90 β plays a critical role during SFTSV infection and could be a potential target for the development of drugs against SFTS.
more >>
SARS-CoV-2 breakthrough infections following inactivated vaccine vaccination induce few neutralizing antibodies against the currently emerging Omicron XBB variants
Highlights
1. Inactivated vaccine breakthrough infection with ancestral or Delta variant induced nearly undetectable nAbs against XBB variants.
2. Inactivated vaccine breakthrough infection with Omicron BA.1 or BA.5 evoked very weak nAbs against XBB variants.
3. BA.5 infection induced higher nAbs against XBB variants than BA.1 infection.
more >>
1. Inactivated vaccine breakthrough infection with ancestral or Delta variant induced nearly undetectable nAbs against XBB variants.
2. Inactivated vaccine breakthrough infection with Omicron BA.1 or BA.5 evoked very weak nAbs against XBB variants.
3. BA.5 infection induced higher nAbs against XBB variants than BA.1 infection.
Drug repurposing screen identifies vidofludimus calcium and pyrazofurin as novel chemical entities for the development of hepatitis E interventions
Hepatitis E virus (HEV) infection can cause severe complications and high mortality, particularly in pregnant women, organ transplant recipients, individuals with pre-existing liver disease and immunosuppressed patients. However, there are still unmet needs for treating chronic HEV infections. Herein, we screened a best-in-class drug repurposing library consisting of 262 drugs/compounds. Upon screening, we identified vidofludimus calcium and pyrazofurin as novel anti-HEV entities. Vidofludimus calcium is the next-generation dihydroorotate dehydrogenase (DHODH) inhibitor in the phase 3 pipeline to treat autoimmune diseases or SARS-CoV-2 infection. Pyrazofurin selectively targets uridine monophosphate synthetase (UMPS). Their anti-HEV effects were further investigated in a range of cell culture models and human liver organoids models with wild type HEV strains and ribavirin treatment failure-associated HEV strains. Encouragingly, both drugs exhibited a sizeable therapeutic window against HEV. For instance, the IC50 value of vidofludimus calcium is 4.6–7.6-fold lower than the current therapeutic doses in patients. Mechanistically, their anti-HEV mode of action depends on the blockage of pyrimidine synthesis. Notably, two drugs robustly inhibited ribavirin treatment failure-associated HEV mutants (Y1320H, G1634R). Their combination with IFN-α resulted in synergistic antiviral activity. In conclusion, we identified vidofludimus calcium and pyrazofurin as potent candidates for the treatment of HEV infections. Based on their antiviral potency, and also the favorable safety profile identified in clinical studies, our study supports the initiation of clinical studies to repurpose these drugs for treating chronic hepatitis E.
more >>
Naturally occurring PAE206K point mutation in 2009 H1N1 pandemic influenza viruses impairs viral replication at high temperatures
The emergence of influenza virus A pandemic H1N1 in April 2009 marked the first pandemic of the 21st century. In this study, we observed significant differences in the polymerase activities of two clinical 2009 H1N1 influenza A virus isolates from Chinese and Japanese patients. Sequence comparison of the three main protein subunits (PB2, PB1, and PA) of the viral RNA-dependent RNA polymerase complex and subsequent mutational analysis revealed that a single amino acid substitution (E206K) was responsible for the observed impaired replication phenotype. Further in vitro experiments showed that presence of PAE206K decreased the replication of influenza A/WSN/33 virus in mammalian cells and a reduction in the virus’s pathogenicity in vivo. Mechanistic studies revealed that PAE206K is a temperature-sensitive mutant associated with the inability to transport PB1–PA complex to the nucleus at high temperature (39.5 ℃). Hence, this naturally occurring variant in the PA protein represents an ideal candidate mutation for the development of live attenuated influenza vaccines.
more >>
Mutual antagonism of mouse-adaptation mutations in HA and PA proteins on H9N2 virus replication
Avian H9N2 viruses have wide host range among the influenza A viruses. However, knowledge of H9N2 mammalian adaptation is limited. To explore the molecular basis of the adaptation to mammals, we performed serial lung passaging of the H9N2 strain A/chicken/Hunan/8.27 YYGK3W3-OC/2018 (3W3) in mice and identified six mutations in the hemagglutinin (HA) and polymerase acidic (PA) proteins. Mutations L226Q, T511I, and A528V of HA were responsible for enhanced pathogenicity and viral replication in mice; notably, HA-L226Q was the key determinant. Mutations T97I, I545V, and S594G of PA contributed to enhanced polymerase activity in mammalian cells and increased viral replication levels in vitro and in vivo. PA-T97I increased viral polymerase activity by accelerating the viral polymerase complex assembly. Our findings revealed that the viral replication was affected by the presence of PA-97I and/or PA-545V in combination with a triple-point HA mutation. Furthermore, the double- and triple-point PA mutations demonstrated antagonistic effect on viral replication when combined with HA-226Q. Notably, any combination of PA mutations, along with double-point HA mutations, resulted in antagonistic effect on viral replication. We also observed antagonism in viral replication between PA-545V and PA-97I, as well as between HA-528V and PA-545V. Our findings demonstrated that several antagonistic mutations in HA and PA proteins affect viral replication, which may contribute to the H9N2 virus adaptation to mice and mammalian cells. These findings can potentially contribute to the monitoring of H9N2 field strains for assessing their potential risk in mammals.
more >>
Pangolin HKU4-related coronaviruses found in greater bamboo bats from southern China
Coronavirus (CoV) spillover originating from game animals, particularly pangolins, is currently a significant concern. Meanwhile, vigilance is urgently needed for coronaviruses carried by bats, which are known as natural reservoirs of many coronaviruses. In this study, we collected 729 anal swabs of 20 different bat species from nine locations in Yunnan and Guangdong provinces, southern China, in 2016 and 2017, and described the molecular characteristics and genetic diversity of alphacoronaviruses (αCoVs) and betacoronaviruses (βCoVs) found in these bats. Using RT-PCR, we identified 58 (8.0%) bat CoVs in nine bat species from six locations. Furthermore, using the Illumina platform, we obtained two representative full-length genomes of the bat CoVs, namely TyRo-CoV-162275 and TyRo-CoV-162269. Sequence analysis showed that TyRo-CoV-162275 shared the highest identity with Malayan pangolin (Manis javanica) HKU4-related coronaviruses (MjHKU4r-CoVs) from Guangxi Province, whereas TyRo-CoV-162269 was closely related to HKU33-CoV discovered in a greater bamboo bat (Tylonycteris robustula) from Guizhou Province. Notably, TyRo-CoV-162275 has a putative furin protease cleavage site in its S protein and is likely to utilize human dipeptidyl peptidase-4 (hDPP4) as a cell-entry receptor, similar to MERS-CoV. To the best of our knowledge, this is the first report of a bat HKU4r-CoV strain containing a furin protease cleavage site. These findings expand our understanding of coronavirus geographic and host distributions.
more >>
Isolation and characterization of spike S2-specific monoclonal antibodies with reactivity to pan-coronaviruses
Highlights
1. Five broad-spectrum mAbs identified from COVID-19 convalescents and vaccinees possessed the ability to recognize spike S2 of pan-coronaviruses.
2. Five mAbs targeted distinct but conserved epitopes on spike S2 of pan-coronaviruses.
3. Three of these five mAbs were competitively bound to the fusion peptide epitopes of coronavirus spike protein.
more >>
1. Five broad-spectrum mAbs identified from COVID-19 convalescents and vaccinees possessed the ability to recognize spike S2 of pan-coronaviruses.
2. Five mAbs targeted distinct but conserved epitopes on spike S2 of pan-coronaviruses.
3. Three of these five mAbs were competitively bound to the fusion peptide epitopes of coronavirus spike protein.
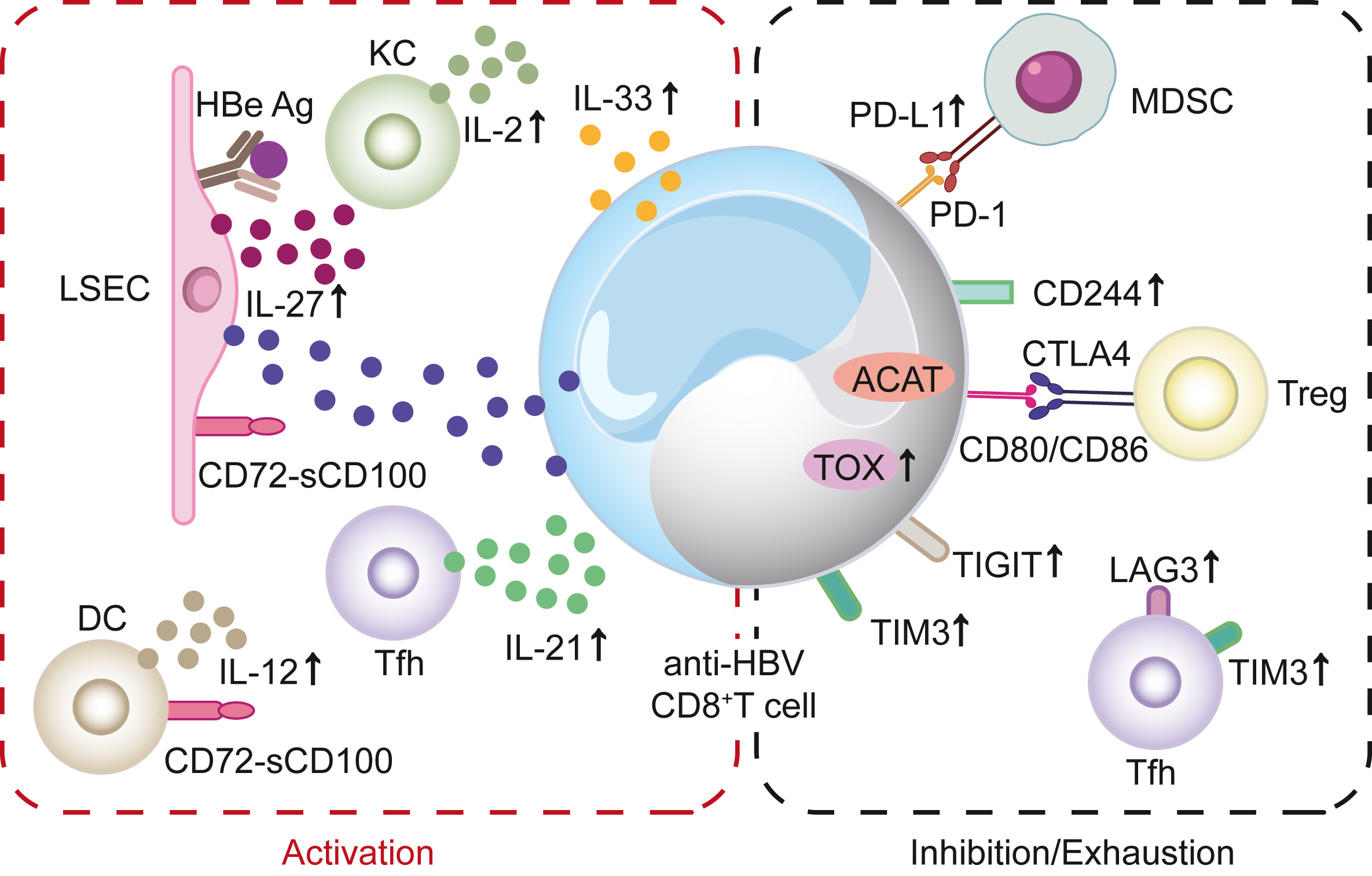
Recent advances in understanding T cell activation and exhaustion during HBV infection
Chronic hepatitis B virus (HBV) infection remains a major public health concern globally, and T cell responses are widely believed to play a pivotal role in mediating HBV clearance. Accordingly, research on the characteristics of HBV-specific T cell responses, from activation to exhaustion, has advanced rapidly. Here, we summarize recent developments in characterizing T cell immunity in HBV infection by reviewing basic and clinical research published in the last five years. We provide a comprehensive summary of the mechanisms that induce effective anti-HBV T cell immunity, as well as the latest developments in understanding T cell dysfunction in chronic HBV infection. Furthermore, we briefly discuss current novel treatment strategies aimed at restoring anti-HBV T cell responses.
more >>
Ebola virus VP35 perturbs type I interferon signaling to facilitate viral replication
As one of the deadliest viruses, Ebola virus (EBOV) causes lethal hemorrhagic fevers in humans and nonhuman primates. The suppression of innate immunity leads to robust systemic virus replication of EBOV, leading to enhanced transmission. However, the mechanism of EBOV-host interaction is not fully understood. Here, we identified multiple dysregulated genes in early stage of EBOV infection through transcriptomic analysis, which are highly clustered to Jak-STAT signaling. EBOV VP35 and VP30 were found to inhibit type I interferon (IFN) signaling. Moreover, exogenous expression of VP35 blocks the phosphorylation of endogenous STAT1, and suppresses nuclear translocation of STAT1. Using serial truncated mutations of VP35, N-terminal 1–220 amino acid residues of VP35 were identified to be essential for blocking on type I IFN signaling. Remarkably, VP35 of EBOV suppresses type I IFN signaling more efficiently than those of Bundibugyo virus (BDBV) and Marburg virus (MARV), resulting in stable replication to facilitate the pathogenesis. Altogether, this study enriches understanding on EBOV evasion of innate immune response, and provides insights into the interplay between filoviruses and host.
more >>
Articles in press have been peer-reviewed and accepted, which are not yet assigned to volumes/issues, but are citable by Digital Object Identifier (DOI).
Column
|
2024, 39(1): 1-8.
doi: 10.1016/j.virs.2023.11.009
Received: 08 March 2023
Accepted: 23 November 2023
Published: 25 November 2023

Single-cell RNA sequencing (scRNA-seq) has allowed for the profiling of host and virus transcripts and host-virus interactions at single-cell resolution. This review summarizes the existing scRNA-seq technologies together with their strengths and weaknesses. The applications of scRNA-seq in various virological studies are discussed in depth, which broaden the understanding of the immune atlas, host-virus interactions, and immune repertoire. scRNA-seq can be widely used for virology in the near future to better understand the pathogenic mechanisms and discover more effective therapeutic strategies.
2024, 39(1): 9-23.
doi: 10.1016/j.virs.2023.12.005
Received: 29 January 2023
Accepted: 13 December 2023
Published: 16 December 2023
The achievement of a functional cure for chronic hepatitis B (CHB) remains limited to a minority of patients treated with currently approved drugs. The primary objective in developing new anti-HBV drugs is to enhance the functional cure rates for CHB. A critical prerequisite for the functional cure of CHB is a substantial reduction, or even eradication of covalently closed circular DNA (cccDNA). Within this context, the changes in cccDNA levels during treatment become as a pivotal concern. We have previously analyzed the factors influencing cccDNA dynamics and introduced a preliminary classification of hepatitis B treatment strategies based on these dynamics. In this review, we employ a systems thinking perspective to elucidate the fundamental aspects of the HBV replication cycle and to rationalize the classification of treatment strategies according to their impact on the dynamic equilibrium of cccDNA. Building upon this foundation, we categorize current anti-HBV strategies into two distinct groups and advocate for their combined use to significantly reduce cccDNA levels within a well-defined timeframe.
2024, 39(1): 24-30.
doi: 10.1016/j.virs.2024.01.001
Received: 07 September 2023
Accepted: 06 January 2024
Published: 09 January 2024
Hepatitis B virus (HBV) produces and releases various particle types, including complete virions, subviral particles with envelope proteins, and naked capsids. Recent studies demonstrate that HBV exploits distinct intracellular membrane trafficking pathways, including the endosomal vesicle trafficking and autophagy pathway, to assemble and release viral and subviral particles. Herein, we summarize the findings about the distinct roles of autophagy and endosomal membrane trafficking and the interaction of both pathways in HBV replication, assembly, and release.
2024, 39(1): 31-43.
doi: 10.1016/j.virs.2023.09.001
Received: 04 May 2023
Accepted: 06 September 2023
Published: 09 September 2023
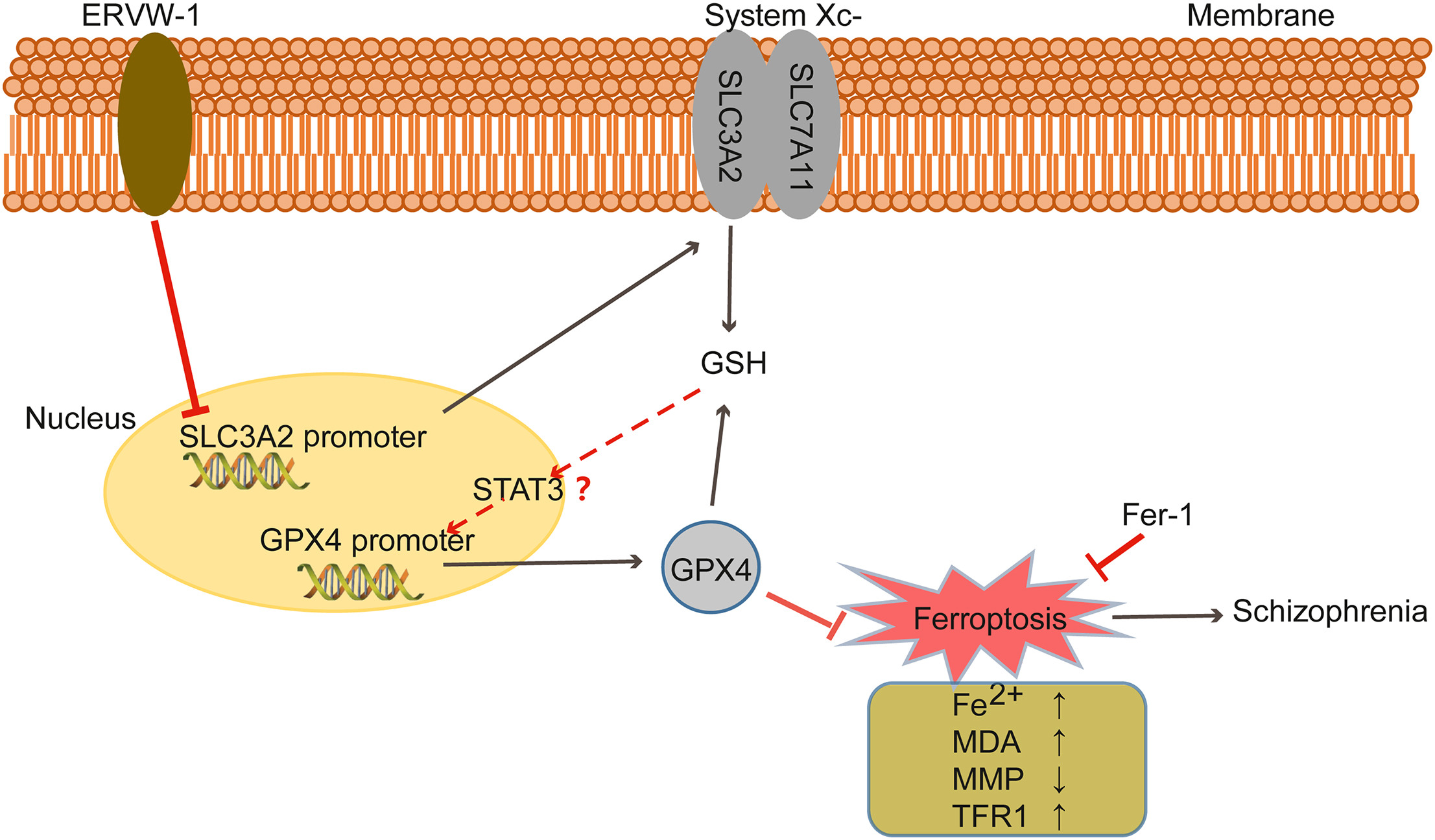
Human endogenous retroviruses (HERVs) are remnants of retroviral infections in human germline cells from millions of years ago. Among these, ERVW-1 (also known as HERV-W-ENV, ERVWE1, or ENVW) encodes the envelope protein of the HERV-W family, which contributes to the pathophysiology of schizophrenia. Additionally, neuropathological studies have revealed cell death and disruption of iron homeostasis in the brains of individuals with schizophrenia. Here, our bioinformatics analysis showed that differentially expressed genes in the human prefrontal cortex RNA microarray dataset (GSE53987) were mainly related to ferroptosis and its associated pathways. Clinical data demonstrated significantly lower expression levels of ferroptosis-related genes, particularly Glutathione peroxidase 4 (GPX4) and solute carrier family 3 member 2 (SLC3A2), in schizophrenia patients compared to normal controls. Further in-depth analyses revealed a significant negative correlation between ERVW-1 expression and the levels of GPX4/SLC3A2 in schizophrenia. Studies indicated that ERVW-1 increased iron levels, malondialdehyde (MDA), and transferrin receptor protein 1 (TFR1) expression while decreasing glutathione (GSH) levels and triggering the loss of mitochondrial membrane potential, suggesting that ERVW-1 can induce ferroptosis. Ongoing research has shown that ERVW-1 reduced the expression of GPX4 and SLC3A2 by inhibiting their promoter activities. Moreover, Ferrostatin-1 (Fer-1), the ferroptosis inhibitor, reversed the iron accumulation and mitochondrial membrane potential loss, as well as restored the expressions of ferroptosis markers GSH, MDA, and TFR1 induced by ERVW-1. In conclusion, ERVW-1 could promote ferroptosis by downregulating the expression of GPX4 and SLC3A2, revealing a novel mechanism by which ERVW-1 contributes to neuronal cell death in schizophrenia.
2024, 39(1): 44-55.
doi: 10.1016/j.virs.2023.10.002
Received: 09 August 2023
Accepted: 08 October 2023
Published: 11 October 2023
Hepatitis B virus (HBV) infection results in liver cirrhosis and hepatocellular carcinoma (HCC). HBx/nuclear factor (NF)-κB pathway plays a role in HBV replication. However, whether NF-κB-interacting long noncoding RNA (NKILA), a suppressor of NF-κB activation, regulates HBV replication remains largely unknown. In this study, gain-and-loss experiments showed that NKILA inhibited HBV replication by inhibiting NF-κB activity. In turn, HBV infection down-regulated NKILA expression. In addition, expression levels of NKILA were lower in the peripheral blood-derived monocytes (PBMCs) of HBV-positive patients than in healthy individuals, which were correlated with HBV viral loads. And a negative correlation between NKILA expression level and HBV viral loads was observed in blood serum from HBV-positive patients. Lower levels of endogenous NKILA were also observed in HepG2 cells expressing a 1.3-fold HBV genome, HBV-infected HepG2-NTCP cells, stable HBV-producing HepG2.2.15 and HepAD38 cells, compared to those HBV-negative cells. Furthermore, HBx was required for NKILA-mediated inhibition on HBV replication. NKILA decreased HBx-induced NF-κB activation by interrupting the interaction between HBx and p65, whereas NKILA mutants lack of essential domains for NF-κB inhibition, lost the ability to inhibit HBV replication. Together, our data demonstrate that NKILA may serve as a suppressor of HBV replication via NF-κB signalling.
2024, 39(1): 56-70.
doi: 10.1016/j.virs.2023.11.004
Received: 14 July 2023
Accepted: 10 November 2023
Published: 13 November 2023
Avian H9N2 viruses have wide host range among the influenza A viruses. However, knowledge of H9N2 mammalian adaptation is limited. To explore the molecular basis of the adaptation to mammals, we performed serial lung passaging of the H9N2 strain A/chicken/Hunan/8.27 YYGK3W3-OC/2018 (3W3) in mice and identified six mutations in the hemagglutinin (HA) and polymerase acidic (PA) proteins. Mutations L226Q, T511I, and A528V of HA were responsible for enhanced pathogenicity and viral replication in mice; notably, HA-L226Q was the key determinant. Mutations T97I, I545V, and S594G of PA contributed to enhanced polymerase activity in mammalian cells and increased viral replication levels in vitro and in vivo. PA-T97I increased viral polymerase activity by accelerating the viral polymerase complex assembly. Our findings revealed that the viral replication was affected by the presence of PA-97I and/or PA-545V in combination with a triple-point HA mutation. Furthermore, the double- and triple-point PA mutations demonstrated antagonistic effect on viral replication when combined with HA-226Q. Notably, any combination of PA mutations, along with double-point HA mutations, resulted in antagonistic effect on viral replication. We also observed antagonism in viral replication between PA-545V and PA-97I, as well as between HA-528V and PA-545V. Our findings demonstrated that several antagonistic mutations in HA and PA proteins affect viral replication, which may contribute to the H9N2 virus adaptation to mice and mammalian cells. These findings can potentially contribute to the monitoring of H9N2 field strains for assessing their potential risk in mammals.
2024, 39(1): 71-80.
doi: 10.1016/j.virs.2023.11.005
Received: 18 July 2023
Accepted: 10 November 2023
Published: 16 November 2023
The emergence of influenza virus A pandemic H1N1 in April 2009 marked the first pandemic of the 21st century. In this study, we observed significant differences in the polymerase activities of two clinical 2009 H1N1 influenza A virus isolates from Chinese and Japanese patients. Sequence comparison of the three main protein subunits (PB2, PB1, and PA) of the viral RNA-dependent RNA polymerase complex and subsequent mutational analysis revealed that a single amino acid substitution (E206K) was responsible for the observed impaired replication phenotype. Further in vitro experiments showed that presence of PAE206K decreased the replication of influenza A/WSN/33 virus in mammalian cells and a reduction in the virus’s pathogenicity in vivo. Mechanistic studies revealed that PAE206K is a temperature-sensitive mutant associated with the inability to transport PB1–PA complex to the nucleus at high temperature (39.5 ℃). Hence, this naturally occurring variant in the PA protein represents an ideal candidate mutation for the development of live attenuated influenza vaccines.
2024, 39(1): 81-96.
doi: 10.1016/j.virs.2023.11.010
Received: 02 February 2023
Accepted: 21 November 2023
Published: 30 November 2023
The mortality of patients with severe pneumonia caused by H1N1 infection is closely related to viral replication and cytokine storm. However, the specific mechanisms triggering virus replication and cytokine storm are still not fully elucidated. Here, we identified hypoxia inducible factor-1α (HIF-1α) as one of the major host molecules that facilitates H1N1 virus replication followed by cytokine storm in alveolar epithelial cells. Specifically, HIF-1α protein expression is upregulated after H1N1 infection. Deficiency of HIF-1α attenuates pulmonary injury, viral replication and cytokine storm in vivo. In addition, viral replication and cytokine storm were inhibited after HIF-1α knockdown in vitro. Mechanistically, the invasion of H1N1 virus into alveolar epithelial cells leads to a shift in glucose metabolism to glycolysis, with rapid production of ATP and lactate. Inhibition of glycolysis significantly suppresses viral replication and inflammatory responses. Further analysis revealed that H1N1-induced HIF-1α can promote the expression of hexokinase 2 (HK2), the key enzyme of glycolysis, and then not only provide energy for the rapid replication of H1N1 virus but also produce lactate, which reduces the accumulation of the MAVS/RIG-I complex and inhibits IFN-α/β production. In conclusion, this study demonstrated that the upregulation of HIF-1α by H1N1 infection augments viral replication and cytokine storm by cellular metabolic reprogramming toward glycolysis mainly through upregulation of HK2, providing a theoretical basis for finding potential targets for the treatment of severe pneumonia caused by H1N1 infection.
2024, 39(1): 97-112.
doi: 10.1016/j.virs.2023.12.003
Received: 01 May 2023
Accepted: 12 December 2023
Published: 14 December 2023
Influenza A virus (IAV) continues to pose a pandemic threat to public health, resulting a high mortality rate annually and during pandemic years. Posttranslational modification of viral protein plays a substantial role in regulating IAV infection. Here, based on immunoprecipitation (IP)-based mass spectrometry (MS) and purified virus-coupled MS, a total of 89 phosphorylation sites distributed among 10 encoded viral proteins of IAV were identified, including 60 novel phosphorylation sites. Additionally, for the first time, we provide evidence that PB2 can also be acetylated at site K187. Notably, the PB2 S181 phosphorylation site was consistently identified in both IP-based MS and purified virus-based MS. Both S181 and K187 are exposed on the surface of the PB2 protein and are highly conserved in various IAV strains, suggesting their fundamental importance in the IAV life cycle. Bioinformatic analysis results demonstrated that S181E/A and K187Q/R mimic mutations do not significantly alter the PB2 protein structure. While continuous phosphorylation mimicked by the PB2 S181E mutation substantially decreases viral fitness in mice, PB2 K187Q mimetic acetylation slightly enhances viral virulence in mice. Mechanistically, PB2 S181E substantially impairs viral polymerase activity and viral replication, remarkably dampens protein stability and nuclear accumulation of PB2, and significantly weakens IAV-induced inflammatory responses. Therefore, our study further enriches the database of phosphorylation and acetylation sites of influenza viral proteins, laying a foundation for subsequent mechanistic studies. Meanwhile, the unraveled antiviral effect of PB2 S181E mimetic phosphorylation may provide a new target for the subsequent study of antiviral drugs.
2024, 39(1): 113-122.
doi: 10.1016/j.virs.2023.11.008
Received: 21 February 2023
Accepted: 22 November 2023
Published: 24 November 2023
Severe fever with thrombocytopenia syndrome (SFTS) caused by the SFTS virus (SFTSV) is an emerging disease in East Asia with a fatality rate of up to 30%. However, the viral-host interaction of SFTSV remains largely unknown. The heat-shock protein 90 (Hsp90) family consists of highly conserved chaperones that fold and remodel proteins and has a broad impact on the infection of many viruses. Here, we showed that Hsp90 is an important host factor involved in SFTSV infection. Hsp90 inhibitors significantly reduced SFTSV replication, viral protein expression, and the formation of inclusion bodies consisting of nonstructural proteins (NSs). Among viral proteins, NSs appeared to be the most reduced when Hsp90 inhibitors were used, and further analysis showed that their translation was affected. Co-immunoprecipitation of NSs with four isomers of Hsp90 showed that Hsp90 β specifically interacted with them. Knockdown of Hsp90 β expression also inhibited replication of SFTSV. These results suggest that Hsp90 β plays a critical role during SFTSV infection and could be a potential target for the development of drugs against SFTS.
2024, 39(1): 123-133.
doi: 10.1016/j.virs.2023.11.006
Received: 05 August 2023
Accepted: 16 November 2023
Published: 19 November 2023
Hepatitis E virus (HEV) infection can cause severe complications and high mortality, particularly in pregnant women, organ transplant recipients, individuals with pre-existing liver disease and immunosuppressed patients. However, there are still unmet needs for treating chronic HEV infections. Herein, we screened a best-in-class drug repurposing library consisting of 262 drugs/compounds. Upon screening, we identified vidofludimus calcium and pyrazofurin as novel anti-HEV entities. Vidofludimus calcium is the next-generation dihydroorotate dehydrogenase (DHODH) inhibitor in the phase 3 pipeline to treat autoimmune diseases or SARS-CoV-2 infection. Pyrazofurin selectively targets uridine monophosphate synthetase (UMPS). Their anti-HEV effects were further investigated in a range of cell culture models and human liver organoids models with wild type HEV strains and ribavirin treatment failure-associated HEV strains. Encouragingly, both drugs exhibited a sizeable therapeutic window against HEV. For instance, the IC50 value of vidofludimus calcium is 4.6–7.6-fold lower than the current therapeutic doses in patients. Mechanistically, their anti-HEV mode of action depends on the blockage of pyrimidine synthesis. Notably, two drugs robustly inhibited ribavirin treatment failure-associated HEV mutants (Y1320H, G1634R). Their combination with IFN-α resulted in synergistic antiviral activity. In conclusion, we identified vidofludimus calcium and pyrazofurin as potent candidates for the treatment of HEV infections. Based on their antiviral potency, and also the favorable safety profile identified in clinical studies, our study supports the initiation of clinical studies to repurpose these drugs for treating chronic hepatitis E.
2024, 39(1): 134-143.
doi: 10.1016/j.virs.2023.12.002
Received: 23 September 2023
Accepted: 04 December 2023
Published: 07 December 2023
The monkeypox virus (MPXV) has triggered a current outbreak globally. Genome sequencing of MPXV and rapid tracing of genetic variants will benefit disease diagnosis and control. It is a significant challenge but necessary to optimize the strategy and application of rapid full-length genome identification and to track variations of MPXV in clinical specimens with low viral loads, as it is one of the DNA viruses with the largest genome and the most AT-biased, and has a significant number of tandem repeats. Here we evaluated the performance of metagenomic and amplicon sequencing techniques, and three sequencing platforms in MPXV genome sequencing based on multiple clinical specimens of five mpox cases in Chinese mainland. We rapidly identified the full-length genome of MPXV with the assembly of accurate tandem repeats in multiple clinical specimens. Amplicon sequencing enables cost-effective and rapid sequencing of clinical specimens to obtain high-quality MPXV genomes. Third-generation sequencing facilitates the assembly of the terminal tandem repeat regions in the monkeypox virus genome and corrects a common misassembly in published sequences. Besides, several intra-host single nucleotide variations were identified in the first imported mpox case. This study offers an evaluation of various strategies aimed at identifying the complete genome of MPXV in clinical specimens. The findings of this study will significantly enhance the surveillance of MPXV.
2024, 39(1): 144-155.
doi: 10.1016/j.virs.2023.12.004
Received: 13 May 2023
Accepted: 13 December 2023
Published: 15 December 2023
Ferroptosis is a newly discovered prototype of programmed cell death (PCD) driven by iron-dependent phospholipid peroxidation accumulation, and it has been linked to numerous organ injuries and degenerative pathologies. Although studies have shown that a variety of cell death processes contribute to JEV-induced neuroinflammation and neuronal injury, there is currently limited research on the specific involvement of ferroptosis. In this study, we explored the neuronal ferroptosis induced by JEV infection in vitro and in vivo. Our results indicated that JEV infection induces neuronal ferroptosis through inhibiting the function of the antioxidant system mediated by glutathione (GSH)/glutathione peroxidase 4 (GPX4), as well as by promoting lipid peroxidation mediated by yes-associated protein 1 (YAP1)/long-chain acyl-CoA synthetase 4 (ACSL4). Further analyses revealed that JEV E and prM proteins function as agonists, inducing ferroptosis. Moreover, we found that treatment with a ferroptosis inhibitor in JEV-infected mice reduces the viral titers and inflammation in the mouse brains, ultimately improving the survival rate of infected mice. In conclusion, our study unveils a critical role of ferroptosis in the pathogenesis of JEV, providing new ideas for the prevention and treatment of viral encephalitis.
2024, 39(1): 156-168.
doi: 10.1016/j.virs.2024.01.006
Received: 08 April 2023
Accepted: 17 January 2024
Published: 20 January 2024
Severe acute respiratory syndrome coronavirus 2 (SARS-CoV-2), the pathogen responsible for coronavirus disease 2019 (COVID-19), continues to evolve, giving rise to more variants and global reinfections. Previous research has demonstrated that barcode segments can effectively and cost-efficiently identify specific species within closely related populations. In this study, we designed and tested RNA barcode segments based on genetic evolutionary relationships to facilitate the efficient and accurate identification of SARS-CoV-2 from extensive virus samples, including human coronaviruses (HCoVs) and SARSr-CoV-2 lineages. Nucleotide sequences sourced from NCBI and GISAID were meticulously selected and curated to construct training sets, encompassing 1733 complete genome sequences of HCoVs and SARSr-CoV-2 lineages. Through genetic-level species testing, we validated the accuracy and reliability of the barcode segments for identifying SARS-CoV-2. Subsequently, 75 main and subordinate species-specific barcode segments for SARS-CoV-2, located in ORF1ab, S, E, ORF7a, and N coding sequences, were intercepted and screened based on single-nucleotide polymorphism sites and weighted scores. Post-testing, these segments exhibited high recall rates (nearly 100%), specificity (almost 30% at the nucleotide level), and precision (100%) performance on identification. They were eventually visualized using one and two-dimensional combined barcodes and deposited in an online database (http://virusbarcodedatabase.top/). The successful integration of barcoding technology in SARS-CoV-2 identification provides valuable insights for future studies involving complete genome sequence polymorphism analysis. Moreover, this cost-effective and efficient identification approach also provides valuable reference for future research endeavors related to virus surveillance.
2024, 39(1): 169-172.
doi: 10.1016/j.virs.2023.10.008
Received: 23 May 2023
Accepted: 24 October 2023
Published: 29 October 2023
Highlights
1. Five broad-spectrum mAbs identified from COVID-19 convalescents and vaccinees possessed the ability to recognize spike S2 of pan-coronaviruses.
2. Five mAbs targeted distinct but conserved epitopes on spike S2 of pan-coronaviruses.
3. Three of these five mAbs were competitively bound to the fusion peptide epitopes of coronavirus spike protein.
1. Five broad-spectrum mAbs identified from COVID-19 convalescents and vaccinees possessed the ability to recognize spike S2 of pan-coronaviruses.
2. Five mAbs targeted distinct but conserved epitopes on spike S2 of pan-coronaviruses.
3. Three of these five mAbs were competitively bound to the fusion peptide epitopes of coronavirus spike protein.
2024, 39(1): 173-176.
doi: 10.1016/j.virs.2023.11.007
Received: 02 August 2023
Accepted: 22 November 2023
Published: 23 November 2023
Highlights
1. Inactivated vaccine breakthrough infection with ancestral or Delta variant induced nearly undetectable nAbs against XBB variants.
2. Inactivated vaccine breakthrough infection with Omicron BA.1 or BA.5 evoked very weak nAbs against XBB variants.
3. BA.5 infection induced higher nAbs against XBB variants than BA.1 infection.
1. Inactivated vaccine breakthrough infection with ancestral or Delta variant induced nearly undetectable nAbs against XBB variants.
2. Inactivated vaccine breakthrough infection with Omicron BA.1 or BA.5 evoked very weak nAbs against XBB variants.
3. BA.5 infection induced higher nAbs against XBB variants than BA.1 infection.