











-
The intricate network of the human immune system comprises the immune organs, immunocytes and lymphatic vessels distributed over almost the entire body. Destruction to the composite elements of this defensive system will undoubtedly cause a variety of disorders. It has been reported that more than half of the T lymphocytes of the human body are in the small intestine (5, 31), making this anatomical site extremely important in studying diseases related to the immune system.
Human immunodeficiency virus type 1 (HIV-1) infection preferentially targets the subset of CD4+ T lymphocytes, which are significantly depleted in the gut-associated lymphoid tissue (GALT) concomitant with the peak of virus replication during the acute phase (19, 35). Later, probably due to the stimulation of acquired immune responses, the viral load drops dramatically to a lower level, which is called the setpoint, and then generally maintains a relatively stable state for months to years depending on genetic background of the host and virulence of the transmitted viral strains. The advent of immunodeficiency is inevitable in most cases. However, the underlying mechanisms driving the process of asymptomatic infection toward an ultimately fatal status remain elusive.
HTML
-
GALT is the largest lymphoid organ in the body. Due to its anatomical location, GALT is continuously exposed to dietary antigens or commensal microor-ganisms. It is believed that a set of relatively perfect mechanisms have been developed by GALT to discriminate harmless materials from dangerous antigens and to ensure efficient nutrient absorption without causing pathogen invasion. An estimated 400 m2 surface area of human gastrointestinal (GI) tract, approximately 200 times larger than that of the human skin, provides an extremely broad digestion platform. Also, GALT has a commensurate and complicated composition infiltrating all over the GI tract, including mesenteric lymph nodes (MLNs), Payer's patches, as well as lymphocytes scattered throughout the intestinal intraepithelium and lamina propria (Fig. 1) (31).
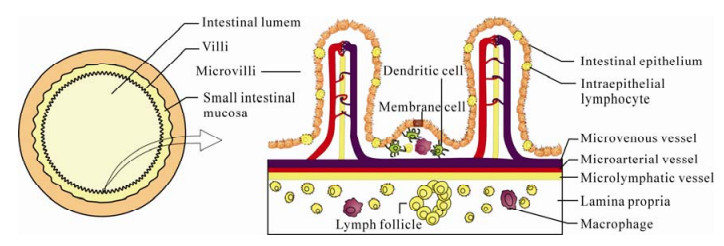
Figure 1. Schematic diagrams of the transverse section of small intestine (left) and intestinal mucosa (right). The villous and microvillous structures enormously broaden the surface area of intestinal mucosa. Abundant memory or activated phenotype lymphocytes are distributed in the mucosal epithelium and lamina propria.
The precise pathway of immune stimulation and lymphocyte migration has yet to be defined. Pathogens are probably taken up by antigen-presenting cells (APCs) situated in the Payer's patches and then delivered through afferent lymphatics to naïve CD4+ T cells in MLNs. Subsequently, primed CD4+ cells up-regulate the expression of chemokines and adhesion molecules (for instances, α4β7, αEβ7, CCR9 and CCR10) which direct these cells homing to effector sites (intraepithelium and lamina propria) (2, 3, 12, 22, 38). More than half of the total T lymphocytes reside in GALT and a considerable part of these cells has an activated or memory phenotype. This abundance of substrate provides a histological basis for massive CD4+ T cell depletion during acute infection because HIV preferentially targets activated CD4+T cells (Fig. 1) (6, 24, 26, 42).
-
Blood contamination, mother-to-child, and sexual contact are the three major routes of HIV transmission of which the latter is particularly important. In the case of mucosal transmission, viral particles or infected cells breach through the mucosal barrier. After local propagation and expansion in the lamina propria underlying cervicovaginal epithelium, viruses disseminate to draining lymph nodes and then start to systemically spread through the circulating system. Within 6-25 days after viral exposure, HIV overcomes the body's initial defenses, disperses to GALT and replicates exponentially, concomitant with fast and massive CD4+ T cell depletion (14). It has been shown that the CD4+ T subset can never recover from such a loss and theirlevels can never be restored to their original values. Even though viral load and peripheral CD4+ cell count can be returned to a near-normal state after long term highly active antiretroviral therapy (HAART), intestinal T lymphocytes remain incom-pletely rehabilitated (10, 14, 16, 17, 40). Deeper impairment to GALT is considered to foreshadow more adverse prognosis and faster disease progression. On the contrary, individuals showing no significant CD4+ T cell loss during acute infection appear to keep virus replication under control and predict slower progression.
-
As reviewed above, the abundance of activated/ memory CD4+ T lymphocytes (CD4+CD25+) residing in GALT provides a histological basis for virus production and CD4+ T cell depletion. Explosive virus replication certainly is a major cause of T cell death during acute HIV infection. However, this is unlikely the whole story because the number of infected cells is observably less than that of lost cells (24). Other by-stander effects (e.g., Fas-Fas-ligand-mediated apoptosis of infected and uninfected cells) are proposed or demonstrated to cause acute CD4+ T cell depletion. In addition, the following causes may also be involved in chronic CD4+ T cell death: (Ⅰ) killing of infected cells by HIV-specific CD8+ T cells; (Ⅱ) antibody-dependent cell-mediated cytotoxicity; (Ⅲ) bystander immune activation and apoptosis of uninfected T cells; (Ⅳ) microbial translocation across the damaged gut mucosa into the circulation; (Ⅴ) compromised thymic function reducing T cell regeneration (15, 28).
-
Natural SIV infections of African nonhuman primates, such as SIVsm infection of sooty mangabeys and SIVcpz infection of chimpanzees, are barely pathogenic (33, 37). These natural infections are generally characterized by the absence of opportunistic pathogen invasion and AIDS-like disease, although there is a marked CD4+ T cell depletion in natural hosts several days after SIV inroad. Several key questions need to be addressed: if massive CD4+ T cell depletion is the fatal causation driving disease progression, why does infection in natural hosts appear to have a benign nature without clinically abnormal manifestations? What is responsible for this markedly different out-come? What are the underlying mechanisms that natural hosts utilize to coexist peacefully with SIV? The answers to these questions may shed light on HIV pathogenesis to a deeper extent and provide potential directions for therapeutic research.
It is well documented that HIV evolved from SIV by crossing the species barriers, although the underlying mechanism remains to be further elucidated. Studies indicate that SIV cross-infection to human happened a few decades ago, indicating that SIV had existed for a very long time before HIV emergence (21, 37). It appears that some yet-to-be-defined strategies have been adapted by nonhuman primate hosts to contain and eliminate the discomforts caused by SIV.
Histological basis
Unrestorable destruction to the immune system
Mechanisms of persistent CD4+ T cell loss
Implications from the nonpathogenic SIV infection of natural hosts
-
Chronic immune activation is marked by non-specific polyclonal B lymphocyte activation, acceleration of T cell turnover, increased frequencies of activated T cells and increased serum levels of proinflammatory cytokines and chemokines (4, 11, 23). Its occurrence strongly suggests the ineluctable onset of AIDS.
-
Rampant replication of HIV during acute phase results in the infection of up to 80% of intestinal memory T cells, which produces severe damage to gut defense system (27). Gut mucosa is a nutrient assimilator, and is also the key barrier to deleterious luminal pathogens. If this important mucosal barrier is disrupted, malabsorption and enteropathy will subsequently occur, explaining the symptom of diarrhea and why HIV infection is a slim disease (29).
HIV-associated immune activation is not fully understood. Nevertheless, opportunistic infection (e.g., pneumocystis jiroveci, Candida species, Cryptococcus, Herpes virus, Cytomegalovirus) and microbial trans-location, ensuing from acute infection, indeed contribute to chronic immune activation (32). Opportunistic pathogens and their components intruding across disrupted intestinal mucosa stimulate innate immunity and create a milieu having markedly elevated proinflammatory cytokines and chemokines. Broad innate immune activation results in acceleration of thymic T cell regeneration and naïve T cell delivery to mucosa sites. This compensatory renewal fuels targets for HIV infection, further promoting the level of specific or non-specific immune activation and gradually exacerbating the problem (8).
Brenchley and colleagues report microbial translocation as the potential etiology of immune activation (9). Their results showed that circulating lipopolysaccharide (LPS) was significantly increased in chronically HIV-infected individuals and SIV-infected rhesus macaques but not in SIV-infected natural host sooty mangabeys. These findings may be attributable to innate immunity. Mandl et al recently revealed that sooty mangabeys have substantially reduced levels of innate immune system activation during acute and chronic SIV infection and that sooty mangabey plasmacytoid dendritic cells produce markedly less interferon-alpha in response to SIV (25). It is not surprising that, LPS, as a microbial product with pathogen-associated molecular patterns recognized by toll-like receptor 4, would induce interferon-alpha production and ultimately result in immune activation (20).
-
There are multiple indicators of the rate of disease progression (summarized in Table 1), although many of them are poorly understood. Chronic immune activation is a stepwise procedure that is presumably present over the entire course of latent infection. In addition to microbial translocation, researchers have proved that Th17 CD4+ T cells, denoting CD4+ T helper cells secreting interleukin 17 (IL-17), play a important role in mucosal immunity (7, 13). Predo-mination of Th1 over Th17 cells or low frequency of Th17 in mucosal sites presages disease progression in SIV infected macaques, likely because of the importance of IL-17 in controlling extracellular bacterial infections. Conversely, unchanged proportions of Th17 were observed in SIV infection of sooty mangabeys.

Table 1. Associated predictors of disease progression
MHC class I tetramers have high binding affinity to corresponding T cell receptors (TCRs) on CD8+ cells. Using highly sensitive assays for specific cytotoxic T lymphocytes (CTLs) responses, researchers have described the key contribution of virus-specific cellular immune responses (especially the CD8+ CTLs) in adaptive immunity. They demonstrate that, if cellular immunity functions properly, virus replication is well controlled; otherwise fast disease progression is most likely heralded. Recent studies found that protective efficacies of CD8+ T cells were represented by polyfunctional profiles capable of producing several cytokines (e.g., IFN-γ, TNF-α, IL-2) simultaneously. In contrast, monofunctionality of CD8+ T subset represents a poor prognosis (1, 34).
Up-regulated expression of programmed death 1 (PD-1) on CD8+ T cells also portends elevated viral loads and disease progression (17, 36, 39). PD-1 is a newly identified member of the CD28 family, expressed on activated CD4+ and CD8+ T lymphocytes, B cells, and macrophages. Interaction of PD-1 with ligands (PD-L1 and PD-L2) expressed on APCs correlates with dysfunction and senescence of CD8+ T cells. Recent studies elucidated that PD-1 was up-regulated on HIV-specific CD8+ T cells in typical disease progressors, but not in long-term nonprogressors, while PD-1 expression was down-regulated in HIV-1 patients with successful response to HAART therapy (41). In addition, blockade of PD-L1 using mAb restored CD8+ cell function.
A small proportion of HIV-infected people, called elite controllers, have the ability to suppress virus replication and maintain normal CD4+ cell count without antiretroviral medications. Another cohort of people, termed long-term nonprogressors or survivors, are capable of staying in latent phase and do not progress to AIDS, although they have suffered immune destruction during acute HIV infection. The underlying mechanism of their resistance to diseases is not well understood. Some studies suggest the roles played by genetic backgrounds, for instance, the presence of HLA-B57 or HLA-B27 allele (18, 30).
Causes of immune activation
Related indicators of disease progression
-
Acute depletion of CD4+ T cells alone, though devastating, is not enough to eventually cause AIDS. Additional pathogenesis is indispensable to drive the process from HIV infection toward AIDS. For instance, CD4+ T cell depletion is observed in natural hosts infected with SIV but does not lead to AIDS. Investigation of SIV natural infection indicates that host adaptability and genetic background, at least in part, play important roles in the manipulation of immunodeficiency virus (19).
Soon after virus exposure, HIV disseminates systematically to GALT and propagates precipitously using the enriched substrates of differentiated CD4+ T lymphocytes, particularly the predominant memory subset. Subsequently, the blunted protective mucosal barrier of the GI tract causes malfunctions (e.g., microbial translocation, Th17 malfunction) and detrimental opportunistic infections. HIV-infected hosts later experience up to several years of asymptomatic phase due to the contributions made by virus-specific adaptive immunity. Viruses mutate and escape constantly throughout this phase even though replicating at a low rate. Chronic immune activation again provides favorable substrate for virus propagation. Poly-to mono-functionality transition and PD-1 up-regulation heralds deteriorated functions of CD8+ cells and accelerated lymphocyte activation. Consequently, the vivus is the winner of the race between HIV mutation and host restoration.
Collectively, there are two times of virus burst in the course of HIV-induced diseases. The first one occurs soon after virus exposure and leads to massive CD4+ T cell loss followed by immune dysfunction and secondary infection of opportunistic pathogens. The second one takes place after chronic immune activation, resulting in incurable AIDS and ultimately death. Massive CD4+ T cell loss provides the prerequisite for chronic immune activation. In the case of HIV infection of elite controllers or/and long-term nonprogressors, AIDS can be prevented (Fig. 2).
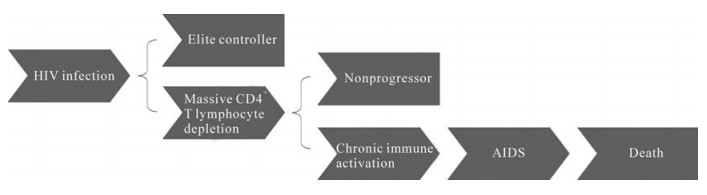
Figure 2. The significance of massive CD4+ T lymphocyte depletion and chronic immune activation during the course of AIDS.
-
With more than 33 million people living with HIV by the end of 2007, HIV/AIDS is undoubtedly one of the most serious public health problems worldwide. This article reviews the two key mechanisms that drive the progression of HIV infection. The most recent findings provide directions for HIV/AIDS control strategies, including prevention of viral replication at mucosal surfaces, restoration of mucosal integrity, blockade of the pathway or reduction of the serum level of microbial translocation, maintenance of normal Th17 frequency, preservation of poly-functionality of HIV-specific CD8+ T lymphocytes and inhibition of ligands interacting with PD-1.

 DownLoad:
DownLoad: