











-
Peste des Petits Ruminants (PPR) is an acute and highly contagious viral disease of small ruminants, characterized by pyrexia, ocular and nasal discharges, erosive stomatitis and diarrhea, and bronchopneumonia (7). It was first described in western Africa in 1940s (6), and has now spread to countries and regions in Sub-Saharan Africa, the Middle East and Asia (12). Epidemiology studies demonstrate a trend for PPR in which it has spread wider and moved eastwards since 1980s (8). The causative agent, Peste des Petits Ruminants Virus (PPRV) has been classified as a member of the Genus Morbillivirus in the Family Paramyxoviridae, containing a single-strand negative sense RNA genome with six genes encoding for eight proteins in the order 3'-N-P/C/V-M-F-H-L-5' (2).
Based on the phylogenetic relationship of the partial F gene sequences, the PPRV isolates have been classified into four lineages (lineages Ⅰ, Ⅱ, Ⅲ and Ⅳ), of which lineages Ⅰ, Ⅱ and Ⅲ were identified in Africa, lineage Ⅲ and Ⅳ were identified in the Middle East, and lineage Ⅳ was identified in Southern Asia. The vaccine virus strain Nigeria 75/1 belongs to lineage Ⅰ and the strain Sungri/96 belongs to lineage Ⅳ (11). Since 1995, several reverse transcription-PCR (RT-PCR) tests have been developed for the rapid and specific detection for PPRV (5), but sequencing remains the gold standard to confirm infection conclusively.
In June, 2007, there was an outbreak of the disease in goats, which first emerged in Ritu County (4), a border county, and which then moved inward to Geji County, Ali, Tibet. A group of experts was organized by the Ministry of Agriculture, PR China, to guide local government for prevention and control of the disease. As a consequence, the outbreak was studied in detail and there were several characteristic findings for the outbreak. Epidemiologically, there was a sudden onset of disease in goat droves without any clinical signs in yaks, a short latent period of around 3 days, an extremely high morbidity rate of up to 100%, and a milder mortality rate of 30-50%. Clinically, higher body temperatures up to 43℃ were observed. Other clinical signs included diarrhea and dehydration, oral, nasal and ocular secretion, and respiratory syndrome in infected goats. Pathologically, lung hemorrhage, necrosis of systemic lymph nodes, and intestinal mucosa hemorrhage were observed. The outbreak was suspected to be caused by PPR based on the fact that PPR outbreaks occur frequently in neighboring countries.
In this study, RT-PCR was used to as a primary diagnostic tool for samples collected from suspected PPR cases in Tibet (4), and positive samples were subsequently confirmed by sequencing.
HTML
-
TaKaRa RNA PCR Kits, TaKaRa One step RNA PCR Kits, pMD18T-vector and DL2000 Markers were purchased from TaKaRa Biotechnology Co., Ltd, Dalian. Gel Extraction Mini kits were purchased from Watson Biotechnologies, Inc, Shanghai.
-
A total of 9 samples numbered from X1 to X9 from sick goats were collected by the local veterinary officers based on clinical and pathological examinations and submitted to the Tibet Center for Animal Disease Control, Lhasa, and 2 other samples (X10 and X11) were collected from sick goats by the expert group during this study. The origins of the tissues are listed in Table 1.

Table 1. The Origin of Tissue Samples
-
RNA samples from the above 11 tissue samples (X1-X11) were prepared in Lhasa, following national biosafety regulations. RNA samples were extracted from collected tissues by employing the TRIzol reagent method (16). Briefly, 1 mL of TRIzol reagent was added to 200 μL of ground tissue suspension, mixed and incubated at room temperature for 10 min. Then 200 μL of chloroform was added, followed by mixing and further incubation at room temperature for 10 min. The solution was centrifuged at 12 000 r/min for 15min in a 220.59 v05 rotor (HERMLE). After collection of the supernatant, equal volume of isopropanol was added, mixed and incubated at -20℃ for 2 h or overnight. The precipitate was collected by centrifugation at 12 000 r/min for 20 min in the same rotor as described above. The pellet was washed with 75% ethanol two times, dried at 37℃ for 8 min, and finally resuspended in 20 μL of RNase-free water. The RNA solution was stored at -70℃ until future use.
-
Primers for detection of F gene and amplification of complete ORF of N/M/F/H genes were designed and synthesized based on previous publications (5, 15). Primers F1 (5'-ATCACAGTGTTAAAGCCTGTAGAGG-3' at position 374-393) and F2 (5'-GAGACTGAGTTTGTGACCTACAAGC-3' at position 783-802) were used for primary F-gene amplification; F1A (5'-ATGCYCTGTCAGTGATAACC-3' at position 802-821) and F2A (5'-YTATGRACAGARGGGACAAG-3' at position 1092-1110) were used for nested amplification of F gene fragments; Nf (5'-ATGGCKACTCTYCTTAAAAG-3') and Nr (5'-GCCGAGGAGATCCTTGT-3'), Mf (5'-ATGACCGAGATCTACG-3'), Mr (5'-CRRGATCTTGAAYRRGCCTTG-3'), Ff (5'-ATGACACGGGTCGCAATCYTG-3'), Fr (5'-CAGTGATCTCACGTACG-3'), Hf (5'-ATGTCCGCACAAAGRGA-3'), Hr (5'-GACTGGATTACATGTTACC-3') were used for the amplification of the complete ORFs of N, M, F, and H genes, respectively.
-
The procedure for RT-PCR and cloning reported in (16) was used for F-fragment analysis as follows: an initial reverse transcription for 45 min at 37℃ was followed by a denaturation step at 95℃ for 5 min. This was followed by 35 cycles of amplification (1 min at 94℃, 1 min at 50℃, and 2 min at 70℃) and a final extension step at 70℃ for 7 min. For amplification of the ORF fragments of N/M/F/H genes, the following was carried out: reverse transcription for 45 min at 42℃, denaturation at 99℃ for 5 min, and then 94℃ for 2 min, followed by 35 cycles of amplification(45 sec at 94℃, 40 sec at 50℃, 2 min at 72℃)and a final extension step at 72℃ for 10 min. The PCR products were analyzed by gel electrophoresis. Known negative and positive samples (Healthy goat tissue and PPRV vaccine strain used as negative and positive control respectively) were included as controls. For cloning, fragments of expected sizes were purified by the DNA purification kit and cloned into the pMD18T-vector following manufacturer's instruction.
-
Products of F gene cDNA obtained with F1A-F2A primers from 5 samples (X2, X5, X8, X10 and X11), and cDNA of OFRs of N/M/F/H genes from Sample 11 were sequenced out and then aligned with published sequences of reference strains using the software package DNAsis version 2.5 (Hitachi, Japan).
Reagents
Sampling
RNA Extraction
Primers
RT-PCR and Cloning
Sequencing, Alignment and phylogenetic analysis
-
A product of approximately 310 base pairs (bp) was detected using the nested F gene primers in all of the 11 samples analyzed in this study. Both negative and positive controls produced results as expected. The results suggested that all 11 samples were positive for PPRV (Fig. 1).
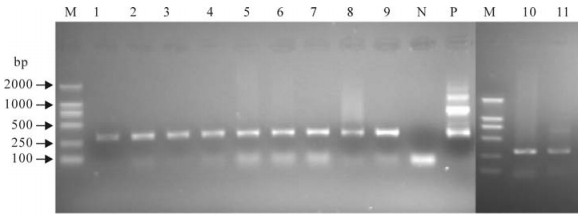
Figure 1. Agarose gel electrophoresis of detected samples amplified with the nested detection primers F1A-F2A. M, DL2000 Marker; N, negative control; P, positive control by using 6 sets of primers simultaneously; Lanes 1-11: samples from X1 to X11 respectively.
-
An alignment of the F gene fragment from 5 samples (X2, X5, X8, X10 and X11) are shown in Fig. 2, with reference strain X74443. A phylogenetic tree based on these sequences was also constructed and is shown in Fig. 3. These results indicated that the PPRV sequences from X2 and X5 were 100% identical and was genetically closest to the strain Nigeria 75/1 (X7443) of lineage Ⅰ. On the other hand, sequences from samples X8, X10 and X11 shared a sequence identity of 98.5% to 99.6% and were most closely related to the strain Sungri96 (AY560591), belonging to lineage Ⅳ.
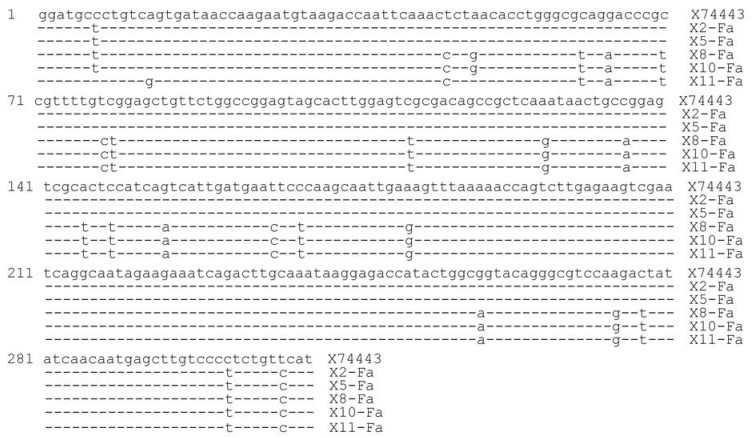
Figure 2. Sequence comparison with reference strain X74443 (GenBank no: GQ184302-06)
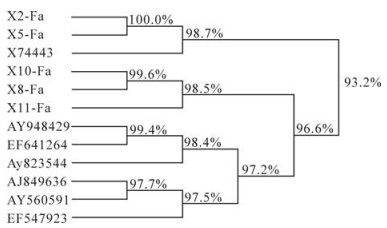
Figure 3. Phylogenetic tree based on the partial sequences of the fusion protein (F) gene.
-
For sample X11, the complete ORF regions for genes N, M, F and H were amplified, cloned and sequenced. The sequences are deposited in GenBank under accession numbers GQ184299-GQ184302, respectively. Sequence comparison and phylogenetic analysis indicated that the X11 sequences were most closely related to the India candidate vaccine virus strain Sungri/96 (AY560591), consistent with previous findings based on the partial F-gene fragment sequences. The sequence identity of X11 and the Sungri/96 strains for N, M, F and H ORFs were 96.6%、97.3%、97.6% and 98%, respectively.
RT-PCR Detection
Sequence Analysis
N/M/F/H ORF Sequence Analysis
-
There is a high risk of PPR being introduced into China from bordering countries and since 2000 several measures have been put in place including an annual serological survey plan, development of diagnostic technologies, risk analysis and establishment of a preventive warning system. In July, 2007, an outbreak of PPR in 2 counties, Ali, Tibet was completely controlled through a series of measures including quarantine, disinfection and slaughtering. Confirmation diagnosis was based on RT-PCR together with direct sequencing of PCR product.
-
Phylogenetic analysis indicated that there were at least two origins of PPRV introduced into China. Several literatures report that PPR has emerged as an endemic disease in India (13), Tajikistan (10) Pakistan and Bangladesh (9), and that there is a growing threat of the emergence of PPR in countries currently free of the disease, especially from PPR endemic areas. Lineage Ⅳ was continuously being reported as circulating in South Asia countries. It is however not clear how lineage Ⅰ in Tibet, China, identified in this study, emerged and its origin remains elusive.
The host range of PPRV was extended to wild small ruminants, but there is no access for Tibetan antelope and yak to the endemic area throughout the history of PPR (3). Much effort has been directed towards investigating the possible source and route of transmission through field investigation. Some evidences implies that movement of domestic livestock may play an important role in transmission of PPR, such as sharing pasture and water source between countries, fosterage among relatives, and the tradition of giving livestock as dowry. However, the exact role of wild ruminants such as Tibetan antelope and yellow goat through its movement or seasonal migration for transmission of PPR still remains unknown. Such study is very difficult, if not impossible, since sample from wild animal is not available due to strict regulation of wild animal protection. However, it is clear that there was no visible clinical sign among wild ruminants during the disease outbreak in Tibet in the regions heavily populated with wild ruminants, especially Tibetan antelope. In our related study, it was shown that a small proportion of domestic yak may be serologically positive without any visible clinical signs, which may also play a role in transmission. (14)
-
Earlier studies have shown similar infection rates in yaks and goats; although the mortality of PPRV infected animals may vary, it can be as high as 100% (1). However, in most of the recent outbreak cases, the mortality is relatively low and is generally less than 50%. The cases of PPR that occurred in the two Counties, Ali, Tibet were mild, with a mortality rate of approximately 25%-35%. Mortality is generally higher in colder areas where animals are exposed to less nutrients than in warmer climate. In our previous study (14), it was shown by PPRV-specific c-ELISA that rate of seropositive animals within a drove of infected goats could be as high as 100%, whereas the seropositive rate within a drove of contact yak without any clinical sign during the outbreak was as low as 11%, e.g. only 1 out of the 9 yaks was serologically positive, which was in striking contrast to a report from Rinderpest regarding a similar outbreak in the same region in the 1950s. However, the reason for this difference is unclear and requires further investigation.

 DownLoad:
DownLoad: