











-
Peste-des-petits-ruminants (PPR) is an acute, highly contagious, Office Internationale des Epizooties (OIE) notifiable, transboundary viral disease of sheep and goats with high morbidity and mortality[11]. The disease is clinically manifested with severe pyrexia, oculo-nasal discharges, necrotizing and erosive stomatitis, enteritis and pneumonia. The etiological agent, PPR virus (PPRV) belongs to the genus Morbillivirus of the family Paramyxoviridae, has a ssRNA genome encoding eight proteins, namely 3' N-P/C/V-M-F-H-L 5', and each gene is separated by intergenic regions[24]. PPR was first reported in the Ivory Coast, West Africa and later from other parts of the world including sub-Saharan Africa, the Arabian Peninsula, the Middle East and parts of Asia[3]. Although, vaccination against PPR is being practiced in India and African countries, PPR still a major constraint in the productivity of small ruminants.
For effective control of the disease, development of an effective prophylactic along with rapid, specific and sensitive diagnostic methods is imperative[11]. Conventional serological methods including virus isolation are time-consuming and insensitive in detecting PPRV during early infection. Monoclonal antibody-based enzyme-immuno assays-ELISA[19, 22, 23] have been used extensively for PPR diagnosis in the mass screening of clinical samples. Earlier, sensitive, reproducible and robust conventional RT-PCR assays based on N, M and F gene sequences of virus have been described for the detection of PPRV[2, 4, 9, 10, 14~16, 21].
Early clinical diagnosis of PPR is difficult due to clinical signs that are confounded with those of other respiratory and enteric diseases, and hence, it needs a laboratory confirmation. The quantitative aspect of the assay is especially important in the diagnosis of sub-clinical and early stages of infection and in particular for quantifying PPRV nucleic acid sequence during pathogenesis studies. Due to the contagious nature of the disease, rapid diagnosis is needed in order to quarantine infected animals and initiate appropriate prophylactic measures. Accordingly, rapid, sensitive, specific diagnostic methods such as RT-PCR assays are reported from various research workers which detect even minute quantities of morbilliviruses or its nucleic acid in the early infection or incubation periods.[4, 5, 12, 13].
The amplification of gene (s) of PPRV in a multiplex or simplex RT-PCR format is usually carried out in two steps, using two reaction mixtures. In the first step, RNA is reverse-transcribed using reverse transcriptase and the resulting cDNA is amplified using DNA polymerase in the second step with two primer pairs. Successful application of the RT-PCR technique depends solely on efficient reverse transcription of the RNA template and subsequent amplification of cDNA by PCR. SYBR green based real time (rt) RT-PCR assays have been applied for diagnosis of many viral diseases of human and animals.
In this study, an earlier available technique is simplified (M gene-based rt TaqMan probe RT-PCR) for rapid detection of PPRV with high sensitivity to allow early diagnosis. The assay was also compared with established s-ELISA[22], M gene-based conventional RT-PCR [2, 16] and hydrolysis probe based rt RT-PCR [4] for efficient detection of PPRV nucleic acid in the clinical materials of sheep and goats.
HTML
-
Attenuated PPR vaccine viruses and cell culture adapted PPRV isolates[3] available in our laboratory were used. Vero cells were grown in Eagle's Minimum Essential Medium (Sigma-Aldrich, St. Louis, MO, USA) supplemented with 10% fetal bovine calf serum for propagation of these viruses.
-
Clinical samples were collected from healthy animals and goats experimentally infected with a goat-adapted virulent challenge PPRV (Izatnagar 94 isolate). These were employed as control and positive samples, respectively. Six-month to one year old, non-descript male goats were experimentally inoculated with 2mL of 10% spleenic suspension of PPRV subcutaneously for normal transmission of the virus to its host (preparation of the virus for PPR vaccine testing is routinely carried out in this laboratory). From this experiment, the nasal, ocular and oral swabs were collected from day 1 to 20 post inoculation (dpi) as preclinical, clinical and post clinical materials from the two goats to assess the virus shedding at early and later stages of the incubation period during pathogenesis of PPR. The infected goats showed typical clinical signs of PPR. Field clinical samples (n=63) of PPRV as infected tissue materials or swabs from different geographical locations in India that were submitted to the laboratory for PPR diagnosis were included for evaluation of assays. Tissue samples were triturated in phosphate buffer saline (PBS-pH 7.2) to prepare a 10% (w/v) suspension, while swabs were directly squeezed or extracted with one 1 mL PBS.
-
Virus RNA was extracted from PPRV purified on a 30–60% discontinuous sucrose density gradient using an RNeasy kit (Qiagen GmbH, Hilden, Germany) according to manufacturer's instructions. The total RNA was extracted from 350 µL of the cell culture adapted different PPRV isolates, homogenates of PPRV-infected materials or swabs from experimentally infected goats and field specimens. To rule out carryover contamination, negative control RNA samples extracted from either mock infected Vero cells or biological samples from healthy sheep and goats were included. QIAamp Blood Mini Kit and QIAamp Viral RNA kit (Qiagen GmbH, Hilden, Germany) were also used to isolate RNA from blood and, swabs and tissue suspension, respectively. The extracted RNA was eluted in 50 µL of nuclease-free water and stored at -20℃ until use. The purity and quantity of the RNA was determined either by Nano drop (Nano Drop 1000, Thermo scientific, Wilmington, DE, USA) or spectrophotometrically. Standard virus (103TCID50) used in rt RT-PCR as control to confirm the successful extraction of RNA. For this, two known negative samples were selected for spiking with a known quantity of PPR vaccine virus and detection of the same by RT-PCR assays.
-
The single-tube one-step rt RT-PCR was standardized using a Brilliant SYBR Green qRT-PCR Master mix kit (M/s Stratagene) using reported M gene-specific primers for PPRV (PPRM Fwd: 5'-TGT G TACATGAGCATAACTAGATTATCA-3';PPRM Rev: 5'-ACTTTCAATTCTTAGTGTAACCAAGATG-3')[4]. The assay was carried out in a 25 µL reaction mixture containing 5 µL of RNA, 10pmoles of each primer, 12.5 µL of 2x SYBR qRT-PCR master mix and 0.0625 µL of StrataScript RT/RNase block enzyme mixture (all the reagents provided in the kit) in a Mx3000p machine (Stratagene Inc., La Jolla, USA). The cycling conditions were as follows: reverse-transcription at 50℃ for 30 min, activation of DNA polymerase at 95℃ for 10 min, 35 cycles of 95℃ for 30 sec, 56℃ for 1 min (to detect and report fluorescence during the annealing/extension step of each cycle) and 72℃ for 30 sec with a final dissociation curve program of 95℃ for 1 min, ramping down (95-55℃) at a rate of 0.2℃ per sec and collecting fluorescence data continuously on the 95-55℃ ramp.
Analytical sensitivity of the assay was determined using PPRV RNA (~500 ng of virus RNA per µL) extracted from purified virus as positive control. Serial ten-fold dilutions of RNA ranging from 50 ng to 0.05 fg (10-1 to 10-10) in 5 μL were tested using one step rt RT-PCR. Standard curve (SC) was generated using serial ten-fold dilutions of virus RNA for absolute quantification as well as to find out the sensitivity and efficiency of the assays. Mean cycle-threshold values of duplicate/triplicate samples were used for analysis. Further, PPRV with a titre of 105TCID50/mL was diluted 10-fold serially from 10-1 and 10-10 and RNA was extracted. Subsequently, RT-PCR reactions were performed as described above to determine the detection limit of the assay. The sensitivity was defined as the lowest quantity of virus RNA giving an amplification signal.
-
The RNA templates were subjected to the conventional RT-PCR utilizing the MF-Morb (5'-CTTGATACTCCCCAGAGATTC-3') and MR PPR 3 (5'-TTCTCCCATGAGCCGACTATGT-3') primers as per the methods described earlier[2, 16]. M gene-based TaqMan rt RT-PCR was also carried out in a Mx3000p qPCR machine (Stratagene Inc., La Jolla, CA, USA) as per the method described earlier[4].
All the clinical samples were tested in parallel in s-ELISA[22], conventional, TaqMan rt RT-PCR and one-step SYBR Green rt RT-PCR assays for analyzing the comparative efficiency of the assays. The one-step SYBR Green assay was applied to the RNA extracted from clinical samples obtained from experimentally infected goats and field specimens (n =63) collected from sheep and goats suspected of PPR infection.
Viruses and cells
Clinical samples
Preparation of RNA
One step SYBR Green rt RT-PCR
RT-PCR assays
-
Real-time PCR is an accurate, rapid and reliable method that can be used for the detection and also for the quantitation of specific DNA molecules. Clinical diagnosis of PPR is difficult as symptoms may be confounded with those of other respiratory and enteric diseases, which should be differentiated at an early stage of infection in suspected cases using confirmatory diagnostic assay. In recent years, several attempts have been made to improve diagnosis of PPR and thereby several RT-PCR assays were developed to allow efficient virus detection in vivo from swabs or biological samples[2, 4, 5]. A two-step RT-PCR with RT and PCR in separate reactions has been widely used for detection of PPRV. The sensitivity, specificity and rapidity of RT-PCR compared with conventional methods, including virus isolation, immunofluorescence and ELISA, render this assay the first choice for diagnostic assay. However, conventional RT-PCR is tech nically demanding and requires 4-8 h for a complete diagnosis compared to 3-4 h in real time PCR for the same results.
The rt RT-PCR assay displays several advantages over the conventional method, such as increasing the laboratory throughput, enabling simultaneous processing of several samples and the ability to quantify the virus load in clinical specimens. Moreover, it gives exhaustive results within 3-4 h and the reaction is performed in a closed-tube, not requiring additional manipulations, which makes the fluorogenic PCR assay useful for pathogenesis studies. A problem with the two step PCR technique is false positives resulting from carryover contamination due to extensive manipulations. The one-step RT-PCR over comes this problem and has been used to detect various pathogens directly in clinical samples[2, 6, 18]. This technique has the advantages of being more sensitive and rapid than the classical one and it also minimizes the chances of carryover contamination[17, 20].
The aim of this study was first to detect the PPRV in the clinical specimens from sheep and goats rapidly with high sensitivity by rt RT-PCR using SYBR green chemistry for early diagnosis and to compare efficacy of this sensitive assay with established assays for efficient detection of PPRV in the clinical materials.
The thermal profile of PCR was optimized to achieve maximum efficiency of amplification. The SYBR green rt RT-PCR was carried out at annealing temperatures (Ta) ranging from 54 to 58℃. The remaining thermo-profile steps were constantly maintained throughout the assay. Of the various Ta that were considered in the rt RT-PCR, a Ta of 56℃ was considered optimum with an amplification efficiency [Y = –3.317 Log X + 13.55] of 100%. The efficiency (10-1/slope) of amplification (100.31) was approximately 2 with an R2 value of 0.998 in the SC (Fig. 1A), indicating the doubling of expected product after each cycle. The assay amplifies 124 bp fragment of PPRV M gene with a Tm of 78.28 to 78.50 (Fig. 1B).
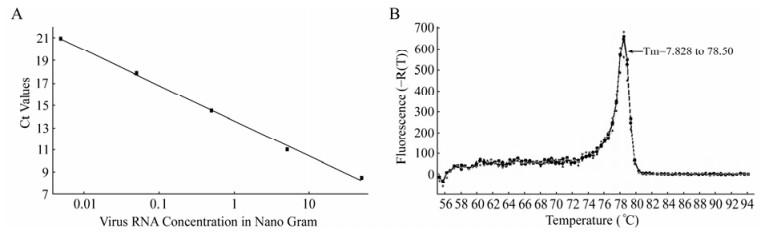
Figure 1. M gene based SYBR green real time RT-PCR. Standard curve obtained with different concentrations of RNA ranging from 50 ng to 5pg. Each dot on the curve represents dilutions of the RNA ranging from 10-1 to 10-5 (A). Analysis of PPRV-specific M gene products by melting dissociation curve showed Tm=78.3 to 78.5℃ (B).
The rt RT-PCR assay was able to detect virus RNA concentration of 0.5fg per reaction mixture, with a corresponding Ct value of 33.8±0.34. The detection limit of conventional RT-PCR was 5 pg of RNA, which was 10-times less sensitive than TaqMan rt RT-PCR (detection limit of 0.5 pg of virus RNA)[4]. The dynamic range of the assay was found to be over a 10-log-unit span of virus RNA concentration, ranging from 50 ng to 0.5 fg of RNA per reaction mixture. The analytical sensitivity of the rt RT-PCR assay was determined through ten serial dilutions of RNA concentration per reaction mixture to generate a SC. The samples that did not yield any Ct value within 35 cycles that were considered negative for PPRV, as the fluorescence remained static or below the base level. The fluorescence remained static even for known positive samples (PPRV), when they were used at higher dilutions indicating the absence of virus-specific amplicons.
Further, to find the detection limit of the rt RT-PCR assay at the TCID50 level, total RNA extracted from a 10-fold dilution of PPR vaccine virus (105TCID50/mL) was tested. The rt RT-PCR assay was able to detect up to 10-9 sample dilution, with a corresponding CT value of 34.6±0.20. At the same time, the analytical sensitivity of the conventional and TaqMan RT-PCR was found to be 10-5 and 10-6 dilution, respectively[4]. The sensitivity of the rt RT-PCR assay was observed to increase by 1000 and 10 times more than that of the TaqMan rt RT-PCR and conventional RT-PCR assays, respectively. Positive and negative controls of clinical samples yielded expected results in the assay. When these samples were tested for comparison by a two-step RT-PCR assays identical results were obtained, reconfirming the suitability of the new assay for the detection of PPRV in infected tissues or swabs.
To determine the detectability and the linearity of the assay (Fig. 2) serial 10-fold dilutions of RNA were used for amplification. The Ct values (y-axis) were measured in three consecutive experiments and were plotted against the known concentration of virus RNA (x-axis). The generated SC covered a linear range of nine orders of magnitude and showed linearity over the entire quantitation range (slope=-3.257), providing an accurate measurement over the range of initial target concentration. The co-efficient of linear regression (R2) was 0.999 with a PCR efficiency of 100 %.
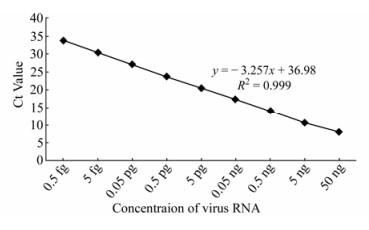
Figure 2. Linear regression curve of the SYBR green real time RT-PCR assay based on ten-fold serial dilutions containing different concentration of standard virus RNA prior to amplification were used, as indicated in the x-axis, where as the corresponding cycle threshold (Ct) values are presented on the y-axis. Each dot represents the result of three consecutive experiments carried out using each dilution. Regression analysis correlation co-efficient (r2) and the slope value (b) are shown.
Finally, to correlate the performance of the developped assay, clinical samples spiked with PPRV were tested as described for MV[1] with some modifications. Two clinical samples (ocular swabs) found negative by M gene based RT-PCR assays in this study, were selected for assessing the exact sensitivity by spiking the known quantity of PPR vaccine virus into the samples and detection of the same by RT-PCR assays. Infected cell culture fluid containing 105TCID50/mL virus was diluted 1:10 serially and spiked 103 and 104TCID50virus in 450 µL of the two negative ocular swabs. The RNA extracted from these spiked samples along with virus controls (103 and 104TCID50virus) was used for RT-PCR reaction, as described earlier in this text.
Mean Ct values of 14.32 ± 0.21 and 17.49.±0.32 were obtained with 102 and 10 TCID50virus samples. Spiked known quantities of PPRV were equally well detected in SYBR green rt RT-PCR assays and the results were identical to those obtained using either conventional or rt RT-PCR assays. The RT-PCR assay for PPRV was reproducible and linear over a range of RNA concentrations or TCID50of virus allowing a precise detection of PPRV RNA load in the sam ples.
The standardized SYBR green rt RT-PCR in the present study using purified virus RNA of PPRV was easily adapted for direct detection of PPRV in clinical field and experimental samples. In the testing of nasal, ocular and oral swabs from goats experimentally infected with PPRV by the SYBR green rt RT-PCR, it was found that the assay could detect the virus in these samples at 3–20 dpi. However, our earlier one step multiplex RT-PCR[2] had detected the virus in swab materials from 5–17 dpi. Samples tested by s-ELISA, detected the virus in preclinical swabs at 7–12 dpi as against 5-15 dpi in conventional RT-PCR and 4-17dpi in TaqMan rt RT-PCR[4]. Using this sensitive assay it may be possible to detect PPRV in preclinical swabs from early stage to a longer course of infection than using s-ELISA. Detectable virus shedding by day 3 or 4 dpi or at least 2-3 days before the clinical signs, was observed by using either RT-PCR or immuno capture ELISA techniques[7, 8], which indicates that incubatory carriers therefore might play a role in the transmission of PPRV among small ruminants. Our results also showed that SYBR green rt RT-PCR has a higher diagnostic efficacy than conventional PCR and s-ELISA for the detection of PPRV in the preclinical samples. Elia et al.[13] also reported that the real time and conventional assays were equally sensitive in the detection of CDV RNA in the clinical samples. Nasal, ocular and oral swabs were found to contain significant virus load and therefore proved to be suitable clinical material for detection of PPRV RNA. Semi-quantitation of virus genetic loads in clinical swab materials collected at different time points after experimental infection showed a good correlation. This finding is consistent with previous results that showed oculo-nasal swabs were good targets for diagnosis of PPR in vivo in clinically suspected sheep and goats[2, 4, 22].
To evaluate the suitability of the SYBR green rt RT-PCR assay for direct detection of PPRV in clinical samples, PPR suspected samples from different geographical regions of India were selected for amplification in the rt RT-PCR and analyzed. The results of testing 63 clinical samples by s-ELISA, conventional RT-PCR, real time TaqMan and SYBR green RT-PCR assays (Table 1) revealed that 18 (28.57%), 35 (55.55%), 49 (77.77%) and 57 (90.47) were positive, respectively. These results indicate that the one step RT-PCR assay is adequate for the detection of PPR viruses from clinical samples. The results also showed higher diagnostic efficacy of the one step rt RT-PCR over the conventional one as it was generally more rapid and more sensitive than the latter. Agreement between the results of the rt RT-PCR assays was very high, indicating the virus load in the clinical samples is sufficient for the detection of the target gene by these assays. False-negative samples by s-ELISA or conventional RT-PCR are eliminated by using the rt RT-PCR assays and were found suitable for the detection of PPRV in clinical samples of various tissues and swabs collected from sheep and goats naturally infected by PPRV.

Table 1. Consolidated results of different assays in the detection of PPRV in the clinical samplesa
The rt RT-PCR assay reported here was found to be rapid (it requires only 3-4 h), sensitive and a suitable screening tool in addition to the earlier available techniques for confirmatory detection of PPR viruses in the clinical specimens of PPR suspect cases. This SYBR green rt RT-PCR assay represents an improvement over earlier methods of PPRV detection due to its rapidity, simplicity, reproducibility and ability to be carried out in laboratories without the need for cell culture facilities. Real-time RT-PCR assay for PPR diagnosis offers substantial improvements in virus detection and thus may aid control of PPR. In India, present clinical surveillance for detection of virus antigen by s-ELISA can be synergistically strengthened with detection of nucleic acid sequences in clinical samples collected from animals, which could be either suspected of having the disease or are at risk of getting the disease. The assay has the potential to be used as a complementary test with conventional assays for diagnosis of PPR and can be useful for diagnostic purposes.

 DownLoad:
DownLoad: