











-
The human immunodeficiency virus (HIV) genome not only contains structural genes, but also a series of accessory genes. The vpu gene is a unique accessory gene to HIV-1 and it encodes a small transmembrane protein [7]. Some studies have shown that Vpu is very important for the maturation and release of HIV-1 [7, 22]. The Vpu-deleted HIV-1 (HIV-1ΔVpu) virions accumulated in the intracellular regions and at the cell surfaces of the HIV-1 infected cells [16]. So the Vpu protein counteracts the effect of inhibitors on HIV-1 virion release. The accelerated effect of HIV-1 release by the protein Vpu is achieved through recruiting cullin-ring finger ubiquitin ligases to eliminate host cell proteins that impede replication and release [13]. In the HIV-1 life cycle, the Vpu protein reduced the cell surface expression of CD4 and recruited cullin1 to efficiently facilitate the release of HIV-1 virons from infected cells [13].
The effect of Vpu is counteracted by the tetherin transmembrane protein [16, 24]. Tetherin is an interferon-induced protein [23, 24], also termed HM1.24 [3], bone marrow stromal antigen 2 (BST2) [6] and cluster of differentiation 317 (CD317) [4]. Tetherin is a raft-associated membrane protein, with a N-terminus cytoplasmic tail, a membrane spanning domain, an extracellular domain, and a C-terminus glycosylpho sphatidylinositol (GPI) membrane anchor [9, 16, 23]. The C-terminal GPI structure enables tetherin localization within cholesterol enriched lipid rafts which is important for HIV-1 release at the plasma membrane [23]. HIV-1 selectively buds from the glycolipid-enriched membrane lipid rafts and HIV-1 virion particles containing the GPI-linked proteins [17]. So it is presumed that tetherin inhibits the HIV-1 release in this way.
Because of the counteractive relationship between Vpu and tetherin, inhibition of Vpu function and mobilization of tetherin's antiviral activity is a potential therapeutic strategy in HIV/AIDS [16]. In this study, Vpu-targeted small interfering RNA (siRNA) that effectively knocks down the expression of the HIV-1 Vpu protein was designed [13, 15]. As a host factor, tetherin that is counteracted by HIV-1 Vpu can also be over-expressed to enhance the antiviral ability of an infected cell [2, 5]. Replication and release of HIV-1 may be inhibited effectively by the combination of Vpu-targeted siRNA and a tetherin over-expressed retrovirus. In this study, both pSIREN-RetroQ-vpuRNAi and pMSCV-tetherin retroviral vector plasmids were packaged into retroviruses in the same packaging cell line. Mixed plasmids were then transfected into the PT67 cells at different ratios, and the mixed retroviruses were harvested. The inhibition efficiency of mixed retroviruses was detected in the TZM-bl cells pre-infected with HIV-1 NL4.3. Then the combined inhibition of vpu RNAi and tetherin over-expression was evaluated based on the individual inhibition effect. Finally, we identified the optimum ratio of mixed retrovirus that achieved the best inhibitory effect on HIV-1 replication.
HTML
-
The HEK293T cell line, derived from transformation of HEK293 cells with the SV40 large T gene [19], was used for the expression of recombinant genes. The PT67 cell line (Cat. No. 631510, Clontech) derived from NIH-3T3 cell contains the moloney murine leukemia virus (MoMuLV) gag, pol, and env (10A1-derived) genes [14] and was used for production of replication-incompetent retrovirus after transfecting with pMSCVneo. The TZM-bl cell line (also called JC.53bl-13) is a HeLa cell derivative that was engineered by amphotropic retroviral transduction and was used to express CD4 and CCR5 and integrated copies of the luciferase and -galactosidase genes under control of the HIV-1 promoter [8, 18]. All of the above cells were cultured in Dulbecco's Modified Eagle Medium (DMEM), supplemented with 10% fetal bovine serum (FBS), 0.1% penicillin, and 0.1% streptomycin at 37 ℃ with 5 % CO2.
-
The plasmid pSIREN-RetroQ was double enzyme digested by Bam H Ⅰ and Eco R Ⅰ. The 6.4 kb fragment was gel purified by a gel extraction kit (DC3511, BIOMIGA). Sequences of Vpu-targeted siRNA were obtained from the online siRNA design tool (http://sivirus.rnai.jp/HIV/) designed by Naito et al. [15]. Target sequences were synthesized as oligonucleotides by Sangon Biotech (Shanghai) and are shown in Table 1. Each oligonucleotide was resuspended in Tris-EDTA (TE) buffer to a concentration of 100 μmol/L and the top strand and the bottom strand of oligonucleotides were mixed at a 1 : 1 ratio up to 30 μL. The mixture was then annealed at 95 ℃ 30 s / 72 ℃ 2 min/ 37 ℃ 2 min/ 25 ℃ 2 min for two cycles. The annealed product was ligated into the purified RNAi-Ready pSIREN-RetroQ vector. Detailed of the protocols are available in the BD™ Knockout RNAi Systems User Manual (PT3739-1).

Table 1. Oligonucleotides used to produce shRNA expression vectors and plasmids construction
The pMSCVneo plasmid was double enzyme digested by Xho Ⅰ and Bgl Ⅱ. The 6.5 kb fragment was gel purified by a gel extraction kit (DC3511, BIOMIGA). The tetherin gene was obtained from the pflag-BST2 plasmid, which is a flag-BST2 fusion protein expressing plasmid, by polymerase chain reaction (PCR). Primers used to amplify tetherin are shown in Table 1. The two enzymes sites Xho Ⅰ and Bgl Ⅱ were added upstream and downstream of the tetherin gene. The tetherin gene was purified with a PCR purification kit (DC3514, BIOMIGA) and ligated into the pMSCVneo vector.
-
In order to get a broad-spectrum inhibition effect on the vpu gene silencing, the vpu gene siRNA target sites should be well conserved. Sequences of Vpu-targeted siRNA were obtained from the online siRNA design tool (http://sivirus.rnai.jp/HIV/) [15]. Five siRNA target sequences were found from 495 HIV-1 vpu sequences which included subtype A, B, C, D, F, G, H, J, K, and are summarized in Table 1. All of these five sequences were annealed and ligated into the purified RNAi-Ready pSIREN-RetroQ vector as described above. To demonstrate whether the siRNA can repress the vpu expression successfully, a vpu-EGFP fusion transcript reporter system was constructed. Primers used to amplify the vpu gene are shown in Table 1. The vpu gene was ligated into the pEGFP-N1 plasmid at the enzyme digestion sites of Eco R Ⅰ and Bam H Ⅰ. The pVpu-EGFP plasmid and pSIREN-RetroQ-vpuRNAi-1~5 plasmids were co-transfected in HEK293T cells (5×105 cells/well on 6 well plates). pSIREN-RetroQ plasmid was used as a negative control and co-transfected with the pVpu-EGFP plasmid. After 48 h, HEK293T cells were observed by fluorescence microscope and images were taken. The RNAi inhibition efficiency was denoted by fluorescence intensity.
-
Tetherin expression of pMSCV-tetherin plasmid was checked by Western blot analysis. The pMSCV-tetherin was transfected into HEK293T cells for the high expression of the recombinant gene. HEK293T cells were seeded in 6 well plates (5×105 cells in each well). 2 μg pMSCV-tetherin plasmid was transfected into cells, and the pMSCVneo plasmid was also transfected as a negative control. After 48 h, cells were harvested for Western blotting assay. Cells were washed twice by 2 mL phosphate buffer solution (PBS) per well, and then cells were detached with 2 mL PBS. The mixture was transferred to a 1.5 mL microtube, centrifuged at 5, 000 rpm for 2 min, and supernatant were discarded. The same volumes of 2x loading buffer (0.1mol/L Tris-HCl pH=6.8, 4% sodium dedecyl sulphate, 20% glycerol, 1% β-Mercaptoethanol, 0.1% bromphenolblue) and 5μL β-Mercaptoethanol were added. Samples were boiled for 15~25min until the solution become clear, centrifuged at 12000 rpm for 5min and supernatants were collected. Supernatants were run on a 10% SDS-polyacrylamide gel along with pre-stained Marker (#SM0671, Fermentas). Blotting, blocking and antibody incubation were performed as previously described in Liu et al. [11, 12], using the mouse polyclonal to BST2 (ab88523, Abcam) at a ratio of 1:1000. Horseradish peroxidase-conjugated goat anti-mouse antibody (AB503, SinoBio) diluted to a ratio of 1:1000 was used as secondary antibody. In order to ensure equal protein loading, a polyclonal anti-β-actin antibody (AB101, SinoBio) was diluted to a ratio of 1:1000 as the primary antibody. Finally, the membrane was treated with western lightning substrates (NEL103001EA, PerKinElmer) using a ChemiDoc XRS imaging system (Bio-Rad).
-
Retroviral particles were packaged in cell line PT67 by transfection of corresponding plasmids. Plasmids pSIREN-RetroQ-vpuRNAi and pMSCV-tetherin were transfected into the PT67 cells (1.2×107 cells in 15 cm dish) using polyethylenimine (PEI). Total plasmids and PEI were mixed for 10 min in the serum-free DMEM with a ratio of 16 μg : 80 μg. Fresh medium was changed at 8 h post transfection. Then 48 h after transfection, the supernatant was harvested and concentrated with ultracentrifugation at 4℃ 32000 rpm for 1.5 h. The supernatant was discarded gently after ultracentrifugation, and 200 µL of serum-free DMEM medium was added into the tube. The pellet was allowed to resuspend at 4℃overnight [20]. Next day, the virus was dispensed and stored at -80℃ for further study.
HIV NL4.3 virus was packaged in cell line HEK293T by transfection of the infectious clone pNL4.3 plasmid [7, 8]. HEK293T cells were transfected with 16 μg pNL4.3 plasmid, supernatant was collected at 60h post transfection. The 50% tissue culture infective dose (TCID50) of virus stock was determined by infecting TZM-bl cells with fourfold serial dilutions of virus [8]. A multiplicity of infection (MOI) was the ratio of TCID50 to cell number [2, 8]. An equal MOI was needed during virus infection in triplicate experiments.
-
Two retrovirus plasmids expressing tetherin and vpu-RNAi were mixed in varying ratios, as shown in Table 2, and then co-transfected into PT67 packaging cells in 15 cm dishes. The detailed process of harvesting virus followed the virus packaging protocol. The harvested mixture of the two retroviruses was then used to infect an indicator cell line TZM-bl in the presence of HIV-1 NL4.3. Inhibition effect was checked in the 96 well plates. The TZM-bl cells were seeded in 96 well plates (4×103 cells in each well) and were infected with HIV-1 NL4.3 with a multiplicity of infection (MOI) of 0.5. After 8 h, the mixed retroviruses at varying ratios were added with a MOI of 0.01. The interference retroviruses mixture was added every 24 h, a total of 4 times, and at 16 h after the last treatment, cells were harvested and luciferase activities were detected to measure the replication of HIV-1. Luciferase assay was according to the rapid protocol for the Steady-Glo assay system (E2520, Promega). The value of relative luciferase activities (RLA) were used to evaluate the effect of the interference retroviruses on HIV-1 NL4.3 replication.

Table 2. Ratios of the two kinds of plasmids used in the virus packaging
Cell culture
pSIREN-RetroQ-vpuRNAi and pMSCV-tetherin plasmids construction
RNAi efficiency assay
Western blotting analysis
Virus packing and MOI determination
Combination inhibition of tetherin over-expression and vpu-RNAi
-
The five vpu-RNAi shRNAs including vpu-shRNA-1, vpu-shRNA-2, vpu-shRNA-3, vpu-shRNA-4 and vpu-shRNA-5 were tested against the Vpu-EGFP fusion mRNA in HEK293T cells. HEK293T cells co-transfected with pVpu-EGFP and pSIREN-RetroQ plasmids served as a positive control, and untreated cells served as a negative control. As shown in Fig. 1, all the five vpu-shRNAs inhibited Vpu-EGFP expression to some extent from 60% to 99%. Vpu-shRNA-3 and vpu-shRNA-4 repressed the vpu gene expression effectively and reached 100% inhibition efficiency. The other three vpu-shRNAs had a lower inhibition effect compared to vpu-shRNA-3 and vpu-shRNA-4. Therefore the vpu-shRNA-3 was selected as the plasmid vector for packing the vpu-RNAi retrovirus.
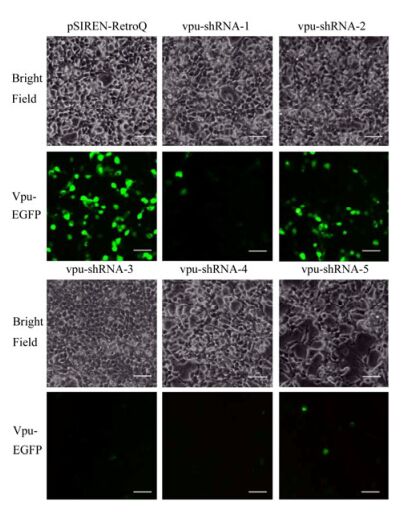
Figure 1. Vpu RNAi efficiency assay. The fluorescence microscopic images of HEK239T cells co-transfected with 1 μg of pVpu-EGFP and 1μg pSIREN-RetroQ plasmids, 1 μg pVpu-EGFP and 1μg pSIREN-RetroQ-vpuRNAi-1~5 plasmids respectively. Fluorescence images were taken at 48 h post transfection (Bar scale=50 μm). Experiments were done in triplicates, and one time images were shown here.
Tetherin over-expression was checked by Western blotting assay. The results suggested that tetherin was over-expressed in HEK293T cells transfected with pMSCV-tetherin, and no expression was observed in the negative control and untreated cells as shown in Fig. 2. When pMSCV-tetherin was transfected into PT67 cells, the tetherin gene was packaged into the retroviral particle. The Tetherin gene could be over-expressed effectively in the cells infected by the tetherin over-expression retrovirus.
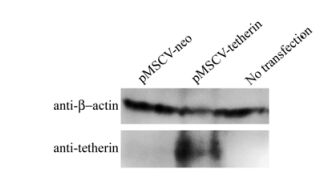
Figure 2. Western blotting assay of tetherin expression in HEK293T cells. After HEK293T cells were transfected for 48 h, they were harvested and a Western blot assay was performed. Experiments were done in triplicates, and one time images were shown here.
-
Relative luciferase activities (RLA) were measured in the TZM-bl cells. The results are shown in Fig. 3. All of the RLA of the experimental group were lower than the HIV-1 NL4.3. So tetherin over-expression retrovirus or vpu-RNAi retrovirus alone or in combination repressed HIV-1 NL4.3 replication in TZM-bl cells in a range from 22% to 57%. Additionally, the inhibitory effect of the vpu-RNAi retrovirus was better than that of the tetherin over-expression retrovirus. The highest inhibition efficiency was observed in the vpu-RNAi virus whereas the lowest therapeutic effect was observed in the combined virus. As expected, inhibition efficiency was reduced with the increase of tetherin. However, this synergistic effect was not observed in the two combined retroviruses of 1T2V and 2T1V, as RLA were reduced by only 55% and 22% respectively. In these cases it appeared that tetherin did not play a role in the combined inhibition the two mixed retroviruses. It is possible that neither the ratio of the two mixed viruses was optimized nor did interfere with the packaging efficiencies of each other during packaging in PT67 cells. Additionally, the vpu gene is highly conserved in the HIV-1 genome [7, 22], and vpu-RNAi can inhibit the vpu gene expression at a rate of 75% (data not published). So the inhibition efficiency decreased along with the quantity of vpu-RNAi viruses in the combined therapy.
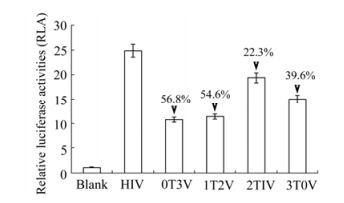
Figure 3. Inhibitory effects of combination infection of tetherin and Vpu-RNAi retrovirus. TZM-bl cells were infected with a MOI of 0.5 in 96 well plates. After 8 h, the mixed retroviruses at varying ratios were added with a MOI of 0.01. The interference retroviruses mixture was added every 24 h, for a total of 4 times. Luciferase activities were assayed on 5 days post infection. The luciferase activity of uninfected TZM-bl cells was regarded as 1, other relative luciferase activities (RLAs) were calculated as divided by the luciferase activity of uninfected TZM-bl cells. Numbers with arrows indicated the decreased extent compared to the non-infection control. Error bars are standard error of triplicate experiments.
The vpu-RNAi expression plasmids repressed the expression of Vpu-EGFP fusion protein and tetherin was over-expressed in HEK293T cells
Packaged combined retroviruses of vpu-RNAi and teherin over-expression inhibited the replication of HIV-1 in TZM-bl cells
-
As a host factor, tetherin inhibits release of the enveloped virus. Meanwhile, tetherin has been shown to be counteracted by the HIV-1 Vpu and HIV-2 Env genes [5, 10, 23]. There are many studies reporting the relationship between HIV-1 Vpu and tetherin [1, 2, 21, 24]. Tetherin inhibits the HIV-1 release [24]. However, HIV-1 Vpu has the ability to down-modulate tetherin from the cell surface [1, 21]. Inhibition of Vpu function or mobilization of tetherin's antiviral activity is a potential therapeutic strategy in HIV/AIDS [16]. We presumed that a combined inhibitory effect is better than a separate one. And in this study, the combined retroviruses of the vpu-RNAi and tetherin over-expression retroviral vector plasmids were packed into the same cell line PT67. In this way it is easy to harvest mixed retroviruses by co-transfecting the packaging cell line. Different ratios of mixed plasmids were checked in order to find an optimized inhibition effect.
In order to get a maximum suppression of HIV-1 vpu gene expression, five vpu-shRNAs were designed. Of these, two were found to repress HIV-1 vpu gene expression effectively and vpu-shRNA-3 was selected as the plasmid vector for packaging the vpu-RNAi retrovirus. According to the RLA value, both individual vpu-RNAi and tetherin over-expressing retroviruses or mixed retroviruses at different ratios could repress the replication of HIV-1 in TZM-bl cells. However, the combined inhibition didn't work any better than the single vpu-RNAi. The vpu-RNAi retroviral vector plasmid and tetherin over-expressing retroviral vector plasmid use the same replicating and packaging factors, so they may compete when co-transfected in the same cell. Considering the mutational escape ability of HIV-1, the individual vpu-RNAi is not practical in HIV-1 gene therapy. However, tetherin is a natural host antiviral factor, so the combined inhibition of vpu-RNAi and tetherin over-expression may provide a novel approach for HIV therapy in the future.

 DownLoad:
DownLoad: