









-
The genus Flavivirus consists of many important arthropod-borne human pathogens such as dengue virus (DENV), Japanese encephalitis virus (JEV), yellow fever virus (YFV), tick-borne encephalitis virus (TBEV) and West Nile virus (WNV) (Gubler D., Kuno, G., Markoff, L., 2007). WNV is widely distributed throughout the world, and is found on all continents except Antarctica (Kramer L D, et al., 2008). Since the initial outbreak of WNV in New York in 1999, WNV has become an important emerging virus. So far, there is no effective antiviral therapy, and no vaccine has been approved for WNV infections.
The WNV genome is a single-stranded, positive-sense RNA molecule approximately 11 kb in length that consists of a 5′ untranslated region (UTR), a single open-reading frame (ORF) and a 3′ UTR. The ORF is translated into a polyprotein that is co-and post-translationally processed by viral and host proteases into three structural proteins (C, prM, and E) and seven nonstructural (NS) proteins (NS1, NS2A, NS2B, NS3, NS4A, NS4B, and NS5) (Brinton M A, 2002; Mukhopadhyay S, et al., 2005). The development of antiviral agents or vaccines is of interest to control WNV infection in addition to mosquito control. However, work with WNV requires biosafety level 3 (BSL-3) facilities, which hinder research for some groups because of safety issues. Previous studies have shown that the complete viral genome RNA without the structural proteins, the replicon, has all the elements for viral replication in transfected cells, but cannot produce infectious viral particles (Shi P-Y, et al., 2002). The replicon system enables researchers to perform studies in biosafety level 2 (BSL-2) facilities. Further study showed that a foreign reporter gene [such as green fluorescent protein (GFP) and Renilla luciferase (Rluc)] could be inserted into the replicon to monitor viral replication (Lo M K, et al., 2003; Shi P-Y, et al., 2002). The Flavivirus replicons are powerful research tools to study the elements of viral replication such as the UTR, nonstructural proteins and viral encapsidation (Lo M K, et al., 2003). The replicon system is also an important tool for antiviral study and drug screening (Alvarez D E, et al., 2005; Holden K L, et al., 2006; Ng C, et al., 2007; Rossi S L, et al., 2005).
In this study, we developed and characterized a WNV replicon (Gluc-WNV-Rep) expressing the secreted Gaussia luciferase (Gluc). The culture medium of Gluc-WNV-Rep RNA-transfected cells was used for a direct assay of luciferase activity to monitor replicon replication. By using a known Flavivirus inhibitor, we showed that GlucWNV-Rep could also be used for antiviral screening.
HTML
-
BHK-21 cells were cultured in Dulbecco's modified Eagle's medium (DMEM; Invitrogen) with 10% FBS, 100 U/mL penicillin and 100 µg/mL streptomycin. For the generation of anti-NS4B anti-serum, BALB/c mice were immunized with a mixture of purified WNV GST-NS4B protein and complete Freund's adjuvant for the primary immunization, and then immunized with two doses of a mixture of purified WNV GST-NS4B protein and incomplete Freund's adjuvant at a 2-week interval.
-
As shown in Fig. 1, an infectious cDNA clone of WNV (designated as pACYC-FLWN), was used to construct a cDNA clone of a Gaussia luciferase WNV replicon (Gluc-WNV-Rep). In the replicon, the coding sequences from 421 to 2379 were replaced with Gluc-2A, while the mature capsid protein and 30 amino acids at the C-terminus of the E protein were retained.

Figure 1. WNV genome and replicons with the Gaussia luciferase reporter constructed in this study. Compared to the full-length WNV genome, the original replicon (Rep) contained an in-frame deletion of the structural region from nt 325 to 2379. The mature capsid protein and the C-terminal 30 amino acids from the envelope protein were retained to maintain the essential 5′ CS element and to ensure correct processing of the polyprotein.
A three-step cloning strategy was used to construct the Gluc-WNV-Rep plasmid. A standard overlap PCR was performed to create a cassette containing the BamHI-T7 promoter-5′ UTR-mature capsid protein, Gaussia luciferase gene, FMDV2A, and the C-terminal 30 amino acids of the E protein to the unique SmaI site in the NS2A protein. The fragment including BamHI to mature capsid was amplified with primer 1 and primer 2 using the WNV infectious clone as a template. The fragment containing the Gaussia luciferase gene was amplified with primer 3 and primer 4 using pGluc-Basic as a template. The fragment spanning the FMDV2A-C-terminal 30 amino acids of the E protein to the unique SmaI site in the NS2A protein (nucleotide position 4018 of the viral genome) was amplified with primer 5 and primer 6 using the pACYC Rluc2A WNV Rep as a template. The primer sequences are presented in Table 1. The three fragments were fused together with primer 1 and primer 6 and then digested by BamH I and Sma I. The fragment from BamH I to SmaI was engineered at the corresponding sites into pACYC-FLWN, resulting in plasmid Gluc-WNV-Rep. A non-replicative NS5 mutation GDD-AAA was introduced into the Gluc-WNV-Rep by site-directed mutagenesis, producing Gluc-WNV-Rep-mt. All the constructs were verified by DNA sequencing.

Table 1. Primers and sequences used in this study
-
The RNAs were in vitro transcribed from XbaI-linearized cDNA plasmids using mMESSAGE MEGAscript® T7 Kit (Ambion) according to the manufacturer's protocols. The RNAs were electroporated into BHK-21 cells as previously described by Zhang B, et al. (2010). After transfection, the supernatants and cell lysates were collected at different time points and used to measure Gluc activity.
-
BHK-21 cells transfected with the Gluc-WNV-Rep and Gluc-WNV-Rep-mt RNAs were seeded on chamber slides (Nalgene Nunc). At 24, 48, and 72 hours post-transfection (h p.t.), the cells were fixed with cold (−20℃) 5% acetic acid in methanol for 15 min at room temperature. The fixed cells were washed with PBS and incubated with anti-WNV-NS4B mouse polyclonal antibody (1:250 dilutions with PBS) for 1 h. After washing with PBS three times, the cells were incubated with goat anti-mouse IgG conjugated to Texas red at room temperature for 1 h. After washing three times with PBS, the slide was mounted with 90% glycerol and examined under a fluorescence microscope (Nikon). Cell images were taken at ×200 magnification.
-
The RNA (10 μg) of Gluc-WNV-Rep was electroporated into 8×106 BHK-21 cells. Electroporation was performed at 25 μF and 850 V with three pulses at 3-s intervals. The cells were seeded in 12-well plates and culture medium was collected at various time points for analysis. The cells were washed once with cold PBS, and 200 μL of lysis buffer (Promega) was added. The plates containing the lysis buffer were sealed with Parafilm and stored at −80℃. When samples had been collected at all time points, 20 μL of cell culture medium and cell lysate were transferred to 96-well plates. Based on the setting procedure, 50 μL BioLux Gaussia luciferase substrate (New England Biolabs, MA, USA) was primed into the sample wells by an injector (Varioskan Flash, Thermo Fisher, Finland), which were then assayed for luciferase signals with a Multimode Microplate Reader (Varioskan Flash).
-
BHK cells transfected with Gluc-WNV-Rep RNA were seeded into 12-well plates at a density of 3.2×105 cells per well. After 2 h, the cells were incubated with various concentrations of NITD008 (0 μmol/L to 27 μmol/L). Incubations were carried out in triplicate, and six control wells were included. The cell culture medium and cell lysates were collected at 48 h.p.t. The Gluc signal was measured with a Multimode Microplate Reader (Varioskan).
Cell lines, antibodies
Plasmid construction
RNA transcription and transfection
Indirect Immunofluorescence Assay (IFA)
Luciferase assay
Inhibition assay of WN-GlucRep by NITD008
-
As shown in Fig. 1, the plasmid of WNV infectious clone pACYC-FLWN was used as a backbone to construct Gluc-WNV-Rep. The structural genes of WNV in the pACYC-FLWN plasmid (Shi P Y, et al., 2002) were replaced with the Gluc reporter gene. We followed a similar strategy for the construction of WNV-Rluc-Rep (Lo M K, et al., 2003; Shi P Y, et al., 2002). In the replicon, the coding sequences of 108 amino acids at the N-terminus of the capsid protein (the mature capsid) and 30 amino acids at the C-terminus of the E protein were retained to preserve a cis element for replication (Khromykh A A, et al., 2001) and the correct processing and translocation of NS1, as well as the correct topology of the remaining nonstructural polyprotein across the membrane of the endoplasmic reticulum. To free the Gluc gene from the polyprotein, the 2A peptide of FMDV was engineered after it. As a negative control, we constructed a mutant counterpart, Gluc-WNV-Rep-mt that contained a frame shift mutation upstream of the active site GDD motif of the RNA-dependent RNA polymerase of NS5 (Fig. 1) (Kim Y G, et al., 2007).
-
We first tested whether Gluc activity could indicate the replication level of Gluc-WNV-Rep. Either Gluc-WNVRep or Gluc-WNV-Rep-mt RNA was transfected into BHK-21 cells. The cell culture medium was collected at 24, 48, and 72 h p.t. and subjected to the luciferase assay. As shown in Fig. 2A, Gluc activity increased dramatically between 24 and 72 h p.t. with Gluc-WNV-Rep RNA. In contrast, transfection with Gluc-WNV-Rep-mt RNA only yielded a background Gluc signal (Fig. 2A). An IFA was used to detect viral protein expression in BHK-21 cells transfected with RNA transcripts. As shown in Fig. 2B, no positive immunofluorescence was detected at 24 h p.t., while an increasing number of WNV-NS4B-positive cells were observed after 48 h p.t. in cells transfected with Gluc-WNV-Rep RNA. No positive immunofluorescence signal was detected in cells transfected with Gluc-WNVRep-mt RNA (Fig. 2B). Taken together, these results demonstrate that the replication of the Gluc-WNV-Rep can be monitored by Gluc activity.
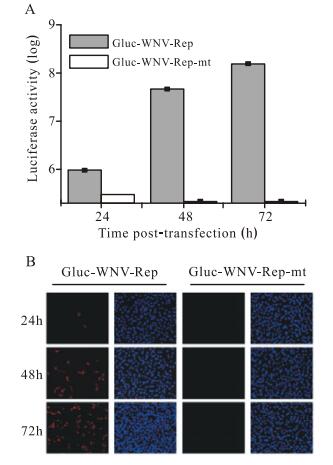
Figure 2. A Gluc reporter can be used to monitor viral RNA replication. A: Gluc activity in cell culture medium at 24, 48, and 72 h p.t. with Gluc-WNV-Rep or Gluc-WNV-Rep-mt; B: IFA of BHK cells transfected with Gluc-WNV-Rep or Gluc-WNV-Rep-mt at the indicated time.
-
To further characterize the Gluc-WNV-Rep system, we transfected BHK cells with equal amounts of Gluc-WNVRep or Gluc-WNV-Rep-mt RNA and monitored Gluc activity at short time intervals within 72 h p.t. Both cell culture medium and cell lysates were collected for luciferase assay. Fig. 3A shows the curves of Gluc activity from cell culture medium. Gluc-WNV-Rep and GlucWNV-Rep-mt produced almost identical curves of Gluc activity until 18 h p.t., demonstrating the equal translation level of the two RNAs. Remarkably, after 18 h p.t., the Gluc signal from cells transfected with the Gluc-WNV -Rep RNA increased exponentially, while the Gluc signal from cells transfected with the Gluc-WNV-Rep-mt RNA remained at the same level from 18 h p.t. to 72 h p.t. Fig. 3B shows the curves of Gluc activity from cell lysates. The signal curves of Gluc-WNV-Rep observed in lysates and culture medium were similar. While for the GlucWNV-Rep-mt, the lysate signal dropped dramatically from 12 h p.t. to 48 h p.t., and then remained at a background level. These results indicated that prior to 10 h p.t., the observed Gluc signal was derived from the translation of the input RNA. After 10 h p.t., the translated Gluc protein began to degrade in the cells, resulting in a decreasing Gluc signal (Tannous B A, 2009). The initially secreted Gluc protein was stable in culture medium, and the Gluc signal could be detected at the same level from 18 h p.t. to 72 h p.t. (Fig. 3A). These results suggest that Gluc signals at early and later time points can be used to quantify the levels of viral translation and RNA replication, respectively.
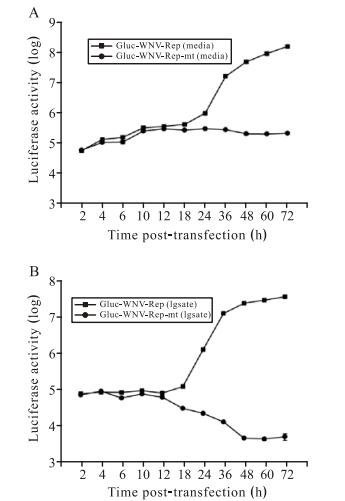
Figure 3. A time course of the Gluc activity in BHK cells transfected with Gluc-WNV-Rep or Gluc-WNV-Rep-mt RNA. A: cell culture medium; B: cell lysate. Similar results were obtained in two independent experiments.
-
To validate whether this Gluc-WNV-Rep could be used as an antiviral screening assay, we used an inhibitor, NITD008 (Yin Z, et al., 2009), which has been proven to be effective against Flaviviruses, to perform the antiviral assay. We examined the inhibitory effects of NITD008 at different concentrations on Gluc-WNV-Rep replication by measuring the luciferase activity of culture medium. As shown in Fig. 4, the Gluc signal decreased with increase in NITD008 concentration (A: medium, B: lysate). The EC50 was estimated to be 0.32µmol/L. These results provided evidence that Gluc-WNV-Rep could be used for screening of WNV inhibitors.
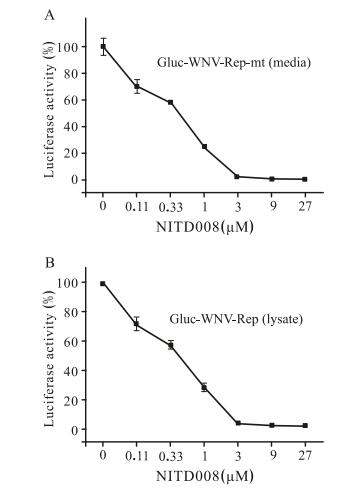
Figure 4. Antiviral activity of NITD008 on Gluc-WNV-Rep. Antiviral activity of NITD008 was assayed using Gluc-WNV -Rep in 12-well plate cultures. BHK cells transfected with GlucWNV-Rep RNA were seeded at a density of 3.2×105 cells per well. After 2 h, the cells were incubated with various concentrations of NITD008 (0μmol/L to 27μmol/L). The cell culture medium (A) and cell lysate (B) were assayed for Gluc signal at 48 h p.t. The data represent three independent experiments performed in duplicate.
Construction of the Gluc-WNV-Rep
Gluc-WNV-Rep could replicate in transfected cells
Gluc-WNV-Rep allowed for quantification and differentiation of translation and replication
Validation of Gluc-WNV-Rep based antiviral screening assay
-
Flavivirus replicon reporter systems are powerful tools for understanding the molecular mechanism of viral life cycles. In this study, we constructed a Gluc-WNV-Rep containing a Gaussia luciferase reporter gene. The Gluc gene is 555 bp, the enzymatic activity is 2, 000-fold higher than that of Renilla luciferase (Tannous B, et al., 2005) and luciferase is secreted into the culture medium of transfected cells. Gluc is naturally secreted from mammalian cells in an active form, and the levels of Gluc in the medium are linear with respect to cell number, growth and proliferation (Badr C E, et al., 2007; Tannous B, et al., 2005). The Gluc reporter system has been shown to be sensitive in monitoring secretory pathways (Suzuki T, et al., 2007), protein–protein interaction (Remy I, et al., 2006), micro RNA (miRNA) biogenesis (Lee J Y, et al., 2008), and promoter activity (Ruecker O, et al., 2008). In vivo, the Gluc reporter system has also been used to monitor tumor growth and proliferation using bioluminescence imaging (Tannous B, et al., 2005; Venisnik K M, et al., 2007). The advantage of the Gluc-WNV-Rep over other reporter replicon systems is that Gluc luciferase activity can be measured in culture media without an additional step of cell lysis. This permits continuous measurement of luciferase activity for monitoring the replication of replicons in a single cell culture. This system could be readily used for high-throughput screening in multi-well culture plates of small molecular mass compounds inhibiting WNV replication.
WNV can cause significant human, equine, and avian disease. Investigation of antiviral strategies to combat WNV is hampered by the requirement of BSL-3 facilities to work with this virus (Kauffman E B, et al., 2011). These biosafety hurdles could be overcome by the use of a WNV replicon system. This system allowed us to rapidly and reliably characterize and quantify WNV translation and RNA synthesis within a BSL-2 facility (Lo M K, et al., 2003; Shi P-Y, et al., 2002). It can provide a rapid and efficient way to understand WNV replication and evaluate candidate antiviral drugs against WNV under BSL-2 conditions.
Overall, our results demonstrate that a rapid, sensitive and efficient replicon system has been developed using Gaussia luciferase as the reporter. The Gluc-WNV-Rep can be used safely under BSL-2 conditions and will be a useful tool for measuring translation and synthesis of WNV RNA, as well as in screening and characterizing anti-WNV compounds without the need of BSL-3 laboratories.
-
We gratefully thank Dr Pei-Yong Shi for assistance during this work. This work was supported by the National Natural Science Foundation of China (grant No. 31170158 and 31000090) and the '100 Talents Project' of Chinese Academy of Sciences, China (grant No. Y002171YC1).

 DownLoad:
DownLoad: