









HTML
-
RNA viruses with either a single-stranded RNA genome (such as influenza viruses belonging to the family Orthomyxoviridae) or double-stranded (ds) RNA segments (e.g. reoviruses or rotaviruses belonging to the family Reoviridae) must possess a virus-encoded, RNA-dependent RNA polymerase (RdRp) responsible for genome transcription and for the replication of their genome. Reoviridae, encompassing 15 genera, is the largest family within the dsRNA virus group (Mertens P, et al., 2011). The virus particles of these viruses show icosahedral symmetry and have an overall diameter of 60-80 nm. The protein shell is organized as one, two or three concentric layers of capsid proteins, which surround 9, 10, 11 or 12 segments of the linear dsRNA genome. In particular, the inner capsids of reoviruses form a transcriptionally active core that acts to protect the viral genome from cellular dsRNA surveillance systems. Thus, reoviruses are an ideal model in which to study mechanisms of RNA-dependent genome synthesis (Mertens P, 2004).
Reoviruses isolated from aquatic animals have been assigned to the genus Aquareovirus, in the family Reoviridae (Mertens PPC, et al., 2011). Grass carp reovirus (GCRV), a pathogen that provokes severe hemorrhagic disease in fingerling and yearling grass carp populations, is regarded as one of the most important pathogens so far identified amongst the aquareoviruses (Rangel A A, et al., 1999). The GCRV particle is a double-layered, spherically structured particle that contains a genome of 11 linear dsRNA segments (named S1-S11) which encode 7 structural proteins (VP1-VP7) and 5 nonstructural proteins (Attoui H, et al., 2002; Cheng L, et al., 2008; Cheng L, et al., 2010; Fang Q, et al., 2005; Fang Q, et al., 2008). Like many other reoviruses, the GCRV particle is organized mainly in two layers: a coat and a core. The coat capsid has an incomplete T = 13 icosahedral structure and is proposed to mediate virus entry by interaction with host cellular factors (Cheng L, et al., 2008; Fang Q, et al., 2005; Li X M, et al., 2013). The inner or core capsid is constructed of proteins VP3 and VP6 and contains the RNA polymerase-related complex (proteins VP1, VP2 and VP4). The core particle is thought to be involved in viral genome replication and transcription. As part of the RNA replicase complex, the VP2 protein of GCRV has been presumed to be a viral RNA polymerase (Cheng L, et al., 2010).
It has been demonstrated that the λ3 protein in mammalian reovirus (MRV) and VP1 in Bluetongue reovirus (BTV) exhibit RdRp activity in the absence of other proteins and cell factors (Boyce M, et al., 2004; Starnes M C, et al., 1993; Wehrfritz J M, et al., 2007). Exceptionally, for rotavirus, the RdRp activity of VP1 is dependent upon the presence of the VP2 protein, which is suspected to be involved in binding the mRNA template (Mansell E A, et al., 1990; Patton J T, et al., 1997). The P2 protein of bacteriophage φ6 is also capable of synthesizing RNA, using both ssRNA and dsRNA as templates, and acts as both a replicase and a transcriptase (Makeyev E V, et al., 2000; Makeyev E V, et al., 2000). In addition, the crystal structure of reovirus RdRp suggests that the RNA polymerases in the Reoviridae family share a common overall configuration, which resembles a cupped "right hand" with three domains of "fingers", "palm", and "thumb" (Ferrer-Orta C, et al., 2006; Lu X, et al., 2008; McDonald S M, et al., 2009; Tao Y, et al., 2002). These RNA polymerase molecules have been identified as being located beneath the pentameric axes of the virus particle; this arrangement facilitates the export of the (+) RNA molecules from the virion core, as in the examples of the mammalian reoviruses, bluetongue virus and rotavirus (Estrozi L F, et al., 2012; Mertens P P, et al., 2004; Zhang X, et al., 2003). In the case of GCRV VP2, as with reovirus RdRps, six conserved structural motifs (denoted A to F) have been recognized, which are supposed to play crucial roles in the catalysis of the nucleotidyltransferase reaction, by binding metal ions, nucleoside triphosphates, RNA and co-factors (Bruenn J A, 2003; McDonald S M, et al., 2009; Tao Y, et al., 2002).
To understand the properties of GCRV VP2, the full-length VP2 gene, with a His-tag fused at its Nterminus, was cloned into a pFastBacTM1 vector and then transformed into DH10BacTM cells. Following transposition, Sf9 cells were transfected with the identified bacmid DNA to produce a recombinant baculovirus expressing VP2 protein. Immunofluorescence (IF) results showed that the recombinant VP2 (rVP2) was stably expressed in Sf9 insect cells. Further analysis by immunoblotting (IB) confirmed the identity and purity of the expressed recombinant VP2 protein. The RdRp activity of the rVP2 recombinants and of viral particles was tested in vitro. The results suggested that GCRV VP2 was capable of poly (C)-dependent poly (G) polymerase activity. The Mg2+ concentration for optimal polymerase activity of VP2 was 10 mmol/L, and the optimal reaction temperature was 28 ℃. The activity could be inhibited by neutralization with VP2 antiserum. Our results will provide the basis for an improved understanding of the molecular mechanisms of GCRV replication.
-
The Ctenopharyngodon idellus kidney (CIK) cell line and the Spodoptera frugiperda Sf9 cell line were used for the propagation of GCRV and of the recombinant baculovirus, respectively. The CIK cells were grown in Eagle's minimum essential medium (MEM, Invitrogen, Grand Island, NY, USA) containing 2 mmol/L L-glutamine, and Sf9 cells were grown in Grace's medium (Gibco BRL, Rockville, MD, USA), supplemented with 10% fetal bovine serum (Gibco BRL, Rockville, MD, USA). The viral strain GCRV-873 was isolated and maintained in the author's laboratory, and the propagation of the virus has been described previously (Fang Q, et al., 1989).
Rabbit polyclonal antibody against GCRV-VP2 protein was generated following the expression of the truncated protein rVP2390-900 in E.coli, as reported previously (Fang Q, et al., 2002). His-tag mouse monoclonal anti-body was the product of Santa Cruz Biotechnology Inc. Alkaline-phosphatase-coupled goat anti-rabbit IgG or alkaline-phosphatase-coupled goat anti-mouse IgG was purchased from Sigma-Aldrich (St. Louis, MO, USA). Alexa Fluor® 568 donkey anti-mouse IgG (H+L) (Red) and Alexa Fluor® 488 donkey anti-rabbit IgG (H+L) (Green) were purchased from Invitrogen Co. (Invitrogen, Carlsbad, CA, USA).
-
To express the VP2 protein in a form convenient for purification, the gene for a recombinant protein with a His-tag fused at the N-terminus was first constructed. The primer pairs were designed based on GenBank sequences (AF260512). The forward primer was 5'-GCTGAATTCATGCATCACCATCACCATCACGAGGAATTGTTCAAC-3' (EcoR Ⅰ site underlined), and the reverse primer was 5'-CATCTGCAGCCACTAAACATCACGCATC-3' (Pst Ⅰ site underlined). The amplified full length of S2 was cloned into a pFastBac-Ⅰ vector that had been cut by EcoR Ⅰ and Pst Ⅰ (TaKaRa, Dalian, China). The positive construct pFbGCRV-VP2 was identified by enzyme digestion and further confirmed by sequence analysis undertaken by Invitrogen Biotechnology Co., Ltd (Shanghai, China). A positive recombinant was used to transform DH10BacTM competent cells (Invitrogen, Carlsbad, CA, USA), which were then grown on SOB medium containing kanamycin, gentamicin, tetracycline, X-gal40 and IPTG. After incubation for 72 h at 37 ℃, white colonies were selected, for the identification of recombinant bacmid DNA (AcGCRV-His-VP2). The isolated recombinant bacmid DNA was verified by PCR with either M13 primers or gene-specific primers, respectively.
-
Monolayers of Sf9 cells were transfected with plasmid DNA from the recombinant bacmid AcGCRV-His-VP2, which was mixed with the cationic liquid Cellfectin reagent, used according to the manufacturer's instructions (Invitrogen, Carlsbad, CA, USA). After 3-5 d post-transfection, the cell suspension was collected by low-speed centrifugation, following the protocol described previously (Fang Q, et al., 2007). The collected supernatant fluid, designated as Passage 1 (P1), was used for the further production of a high-titer P2 stock. The empty bacmid was used as a negative control. To produce stable vAcGCRV-His-VP2 budding virus on a large scale, Sf9 cells were infected with passaged virus stocks and harvested at 72 h post infection (p.i.). The infected cells were washed three times with cold phosphate-buffered saline (PBS), treated with lysis buffer (20 mmol/L Tris-HCl, pH 7.5, containing 150 mmol/L NaCl and 1% [wt/vol] Triton X-100) and then stirred at 4 ℃ for 10 min, prior to three cycles of freeze-thaw. For the analysis of secreted expressed protein, the cell lysate was ultracentrifuged at 80, 000 × g in an SW28 rotor (Beckman) for 2 h at 4 ℃ and the protein pellet was resuspended. Samples of both the cell lysate and the pellet were stored at -70 ℃ for further use. The purification of the His-tag fusion protein was undertaken using a Ni2+-chelating resin column according to the ProBondTM Resin kit manufac-turer's instructions (Invitrogen, Carlsbad, CA, USA).
-
The CIK cells were infected with GCRV at a multiplicity of infection (MOI) of 2 PFU/cell, as described previously (Yan L, et al., 2012). Briefly, the virus was allowed to adsorb to the cells for 1 h and the cells were then washed with PBS to remove the un-adsorbed inoculum. Fresh medium supplemented with 2% of fetal bovine serum (MEM-2) was then added for viral propagation. The virus-infected cell supernatants were collected until such time as the cytopathic effects (CPE) were visible. The GCRV particles were purified using CsCl density gradient centrifugation (Fang Q, et al., 2008). The concentrations of purified recombinant protein and virus particles were measured using a spectrophotometer (Biotek, Wi-nooski, VT, USA). Purified virions were subjected to three cycles of freezethaw before storage at -30 ℃ for further use.
-
All the samples were subjected to 8% SDS-PAGE electrophoresis and transferred to a polyvinylidene fluoride (PVDF) membrane by semi-dry transfer following the manufacturer's directions (Bio-Rad, Hercules, CA, USA). A polyclonal antibody to VP2 was used as the primary antibody and this was followed by incubation with alkaline phosphatase-conjugated goat anti-mouse immunoglobulin G (IgG) or alkaline phosphataseconjugated goat anti-rabbit IgG. All the samples were visualized after development with nitrobluetetrazolium (NBT)/5-bromo, 4-chloro, 3-indolylphosphate (BCIP) alkaline phos-phatase (AP) substrate solution..
IF assays were carried out according to an established method (Shao L, et al., 2013). Briefly, infected or mockinfected Sf9 cells were fixed, made permeable with 0.1% Triton X-100 for 5 min and then blocked with His-tag and VP2-specific antibodies diluted in PBS-BSA, following incubation with bovine serum albumin (PBS-BSA) for 1 h at 37 ℃. After three washes with PBS, all samples were incubated with Alexa Fluor® 568 donkey anti-mouse IgG (H+L) (Red) and Alexa Fluor® 488 donkey anti-rabbit IgG (H+L) (Green). Hoechst staining was applied for the staining of the cell nucleus, and the immunostained cells were observed and the data processed as previously reported (Wen D, et al., 2013).
-
RdRp activity was assayed by assessing the incorporation of radio-labeled nucleotides into a newly synthesized RNA molecule. Unless specifically indicated otherwise, this assay was performed at 28 ℃ in a total reaction volume of 25 μL, containing 50 mmol/L Tris-HCl (pH 7.6), 20 μmol/L GTP, 4 mmol/L DTT, 5 mmol/L MgCl2, 1 μg of Poly (C)/oligo (G) (Amersham Pharmacia Biotech Inc, Piscataway, NJ, USA), 20 units of RNase Inhibitor (TaKaRa, Dalian, China), 5 μCi of [a-32P]-GTP (3000 Ci/mmol, PerkinElmer, Waltham, Massachusetts, USA), and 5 μg/mL actinomycin D (Sigma, St. Louis, MO, USA) to inhibit any possible DNA-dependent RNA polymerase activity. The reaction was initiated by adding 100 ng of purified rVP2; meanwhile, the purified GCRV and rVP2390-900 samples (100ng in each case) were prepared as controls. After incubation for 4 h, 100 μg of salmon sperm DNA (Invitrogen, Grand Island, NY, USA) and 1 mL of 10% [wt/vol] TCA were added to the reaction mixture, which was then left to stand for 30 min at 4 ℃. The mixture was then transferred onto a DE81 Whatman filter (Whatman, Piscataway, NJ, USA), washed twice with 500 mmol/L Na2HPO4 (pH 7.5) and air-dried, prior to the measurement of radioactivity in a scintillation counter (PerkinElmer, Waltham, Massachusetts, USA).
Cell lines, virus and antibodies
Construction and identification of recombinant bacmid
Transfection and recovery of recombinant virus
GCRV Infection and viral particle preparation
Immunoblotting and immunofluorescence analyses
RNA-dependent RNA polymerase (RdRp) assay
-
In order to express the VP2 protein in vitro, the fulllength open reading frame (ORF) of VP2, with the addition of a His-tag sequence designed and incorporated into the N-terminus, was inserted into a pFastBac-1 vector, downstream of the polyhedrin (ph) promoter (as shown schematically in Figure 1A). The recombinant pFbGCRV-VP2 plasmid was purified and used to transform DH10Bac competent cells for transposition. The recombinant bacmid AcGCRV-His-VP2 was identified by PCR, using either M13 primers or gene-specific primers. As shown in Figure 1B, three specific bands with molecular weights of around 6.0 kb were identified in the recombinant bacmid by PCR amplification. Only a 0.3kb band was amplified in the empty bacmid. This result suggested that the VP2 gene had been transposed successfully into the recombinant bacmid.
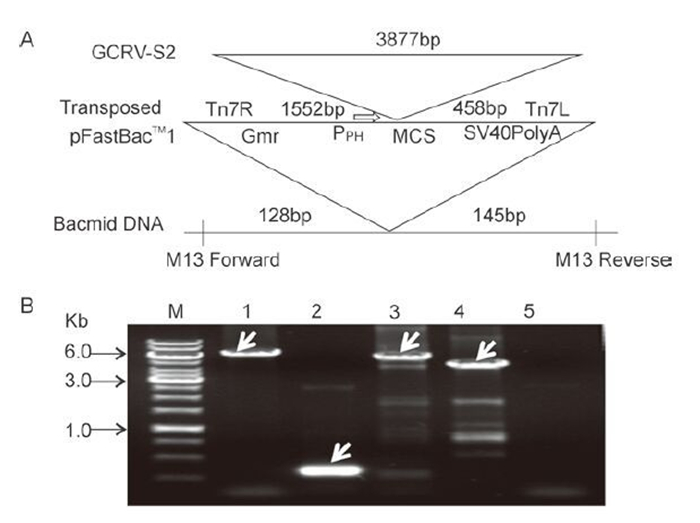
Figure 1. Construction and identification of recombinant AcGCRV-His-VP2 bacmid. A: Schematic representation of the construction of the AcGCRV-His-VP2 bacmid. B: Identification of recombinant AcGCRV-His-VP2. M, 1kb DNA gene ruler; 1, positive amplification with M13-S+M13-AS (~2.3+3.8kb); 2, empty bacmid amplification with M13-S+ M13-AS (~0.3kb); 3 and 4, positive amplification using M13-S+GCRV/S2-AS (~1.7+3.8kb) and GCRV/S2-S+M13-AS (~3.8+0.6kb) primer pairs, respectively; 5, ddH2O control, amplification with M13-S+M13-AS.
-
Sf9 cells were transfected with the recombinant bacmid AcGCRV-His-VP2 to produce recombinant baculovirus passage 1 (P1). P1 stock was clarified by centrifugation and used to infect Sf9 cells at an MOI of 2.5 to generate a high-titer baculovirus stock (P2-P5). Visualization of the intracellular distribution of VP2 was performed by immune-staining with His-tag monoclonal and anti-VP2 polyclonal antibodies, using fluorescence microscopy. Analysis of infected cells revealed that His-VP2 could be detected at 24 h p.i. As shown in Figure 2A, at 24 h p.i. the expression of His-VP2 appeared as small spots of immunofluorescence that were scattered within the infected cells. As infection progressed, at 48 h and 72 h, many large immunofluorescent spots were observed. At 72-96 h p.i., infected cell suspension cultures were collected for the identification of secreted VP2, by IB analysis using anti-VP2 polyclonal antibody. The IB results showed that the specific target band of about 140 kDa, corresponding to the molecular weight of the VP2 protein, was detected in both recombinant baculovirus-infected Sf9 cell lysates and in pelleted supernatant samples (Figure 2B). No such band was detected in mock-infected cells. The IF and IB results therefore indicated that the recombinant protein of interest was correctly expressed in Sf9 cells and that, following expression, the recombinant VP2 protein was secreted into the medium.
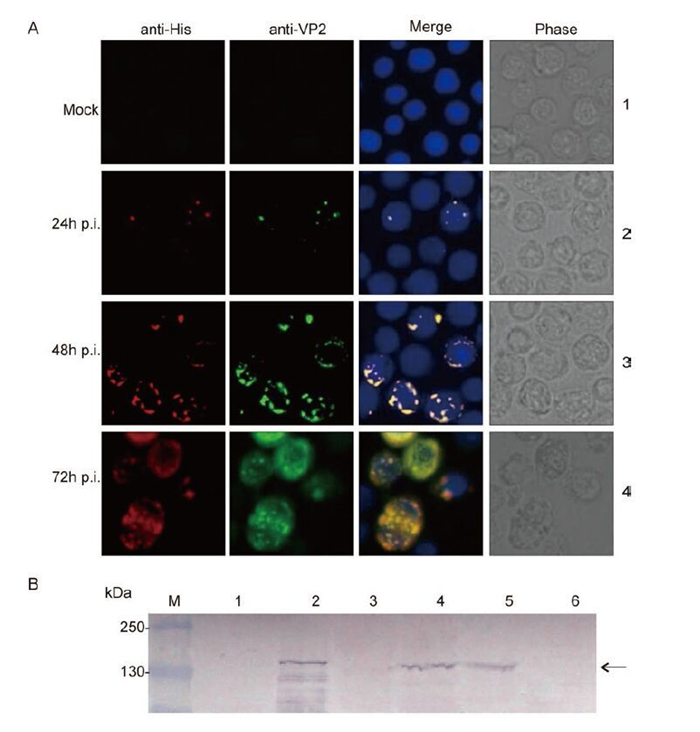
Figure 2. Identification of His-VP2 protein expression in Sf9 cells. A: Mock-infected Sf9 monolayers and Sf9 monolayers infected with a recombinant baculovirus were fixed at 24, 48 and 72 h p.i. and co-immunostained with His-tag monoclonal and VP2 polyclonal antibodies, followed by immunostaining with Alexa 568 anti-mouse IgG (Red) and Alexa 488 anti-rabbit IgG (Green); the nuclei were then counterstained with Hoechst stain. B: Immunoblotting analysis of VP2 expression, using antibody to VP2. 1 and 2, mock-infected and infected Sf9 cell lysate, respectively; 3 and 4, mock-infected and infected Sf9 culture supernatant pellet, respectively; 5 and 6, infected and mock-infected CIK cell lysate.
-
To investigate the function of GCRV VP2 in the absence of other viral or cellular proteins, the soluble His-tagged VP2 was purified by using a Ni2+-chelating resin column. We also prepared purified GCRV particles and rVP2390-900 as controls. As shown in Figure 3, rVP2 was purified and compared both to GCRV particle components and to rVP2390-900. Protein bands of 140 kDa were visualized by SDS-PAGE and Coomassieblue staining for both the purified GCRV and AcGCRVHis-VP2 samples, in contrast to the 55 kDa protein band observed for the rVP2390-900 positive control (Figure 3A). IB assays using VP2-specific polyclonal antiserum further confirmed these SDS-PAGE results (Figure 3B). In addition, the image from the highly purified GCRV particle demonstrated that cell components were absent from the virion sample (Figure 3C). Consequently, all the purified VP2-related samples were suitable for further replicase assays.
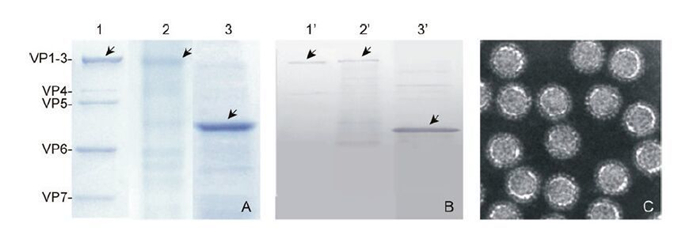
Figure 3. Purification of His-tagged recombinant proteins and virus particles. A: Purified virus particles, rVP2 and rVP2390-900 (shown in lanes 1-3) were subjected to SDS-PAGE, in each case loading 10 μL of sample onto the gel. B: Immunoblot analysis of virus particles and purified rVP2 protein, using antibody to VP2; lanes 1'-3' correspond to lanes 1-3 in A. C: Negatively stained electron micrographs of purified virus particle.
-
As a first step towards establishing an assay system for rVP2, we initially used the poly (C)-dependent, oligo (G)-primed poly (G) polymerase assay (Starnes M C, et al., 1993). In the standard polymerase assay, the enzymatic activity of rVP2 is quantified as the rate of incorporation of [a-32P]-GTP into precipitable material in response to a poly (C) template and an oligo(G) primer. Besides assaying rVP2, we also assayed rVP2390-900 and purified GCRV particles. As shown in Table 1, using poly (C)/ oligo (G) 12-18 as template, there was incorporation of [a-32P]-GTP into a newly synthesized RNA with either rVP2 or GCRV. However, this incorporation was decreased by adding VP2 antiserum to the rVP2 or GCRV reactions (Table 1), indicating that the catalytic activity of the RNA polymerase was inhibited by specific VP2 antiserum. In addition, the incorporation of [a-32P]-GTP was dependent upon the presence of poly (C), because no [a-32P]-GTP incorporation was detected in the absence of poly (C) template (data not shown). No significant incorporation was detected with the purified rVP2390-900.
Sample
(100ng/each)Template/primer 32P-GTP incorporation
(cpm 10-3)Virion Poly(C)/Oligo(G)
Poly(C)/Oligo(G)+VP2 serum21.54
11.70rVP2 Poly(C)/Oligo(G)
Poly(C)/Oligo(G)+VP2 serum16.06
4.35rVP2390-900 Poly(C)/Oligo(G) 0.74 Table 1. Poly (G) polymerase activity assays
The replicase activity of rVP2 was characterized further by investigating the effects of temperature and of the concentration of the divalent cation, Mg2+. The optimal reaction temperature for the incorporation of [a-32P]-GTP into the poly (C) template was 28 ℃ (Figure 4A). Polymerase activity increased gradually as the Mg2+ concentration was raised from 1mmol/L to 10 mmol/L, but was largely inhibited when the concentration was increased further to 50 mmol/L (Figure 4B).
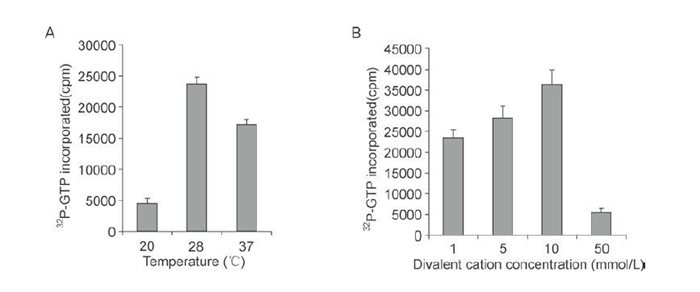
Figure 4. Characterization of RNA polymerase of rVP2. RdRp reactions with 100 ng of purified rVP2 were carried out as detailed in Materials and Methods. Radioactivity was measured by scintillation counting. A: Effect of temperature on replicase activity. B: Effect of Mg2+ concentration on replicase activity.
Identification of recombinant AcGCRV-His-VP2 bacmid
Identification of His-VP2 protein expression in infected Sf9 cells
Purification of His-tag recombinant proteins and virus particles
Enzymic activity of recombinant VP2
-
One of the most unusual characteristics of the dsRNA viruses is that they retain their genomes and mRNA synthesis-related enzymes within a stable and contained shell so as to avoid digestion by host cellular factors (Boyce M, et al., 2004). As with other reoviruses, genome replication and transcription in GCRV also take place within the core particle, through the action of replicase complexes (Mertens P, et al., 2004). The results described in this study suggest that rVP2 and GCRV VP2 each possess poly (C)-dependent poly (G) RNA polymerase activity.
Bioinformatic analysis indicates the presence of several conserved domains within the VP2 protein, such as the motifs SG and GDD that are necessary for RdRp activity (Fang Q, et al., 2002). In addition, there is a high degree of amino acid sequence identity (about 41%) between the putative, RdRp λ3 protein of the orthoreovirus and VP2 protein of the aquareovirus. This suggests strongly that the VP2 protein of GCRV might perform the same role as the λ3 protein of MRV during virus RNA transcription and replication (Fang Q, et al., 2000; Kim J, et al., 2004; Xu W, et al., 2009). To understand the RdRp activity of the GCRV VP2 protein, full-length VP2 was expressed in a baculovirus expression system. It was demonstrated that the purified, amino-terminal His-tagged rVP2 could be recognized by a VP2-specific an-tibody. Further assays of the replicase activity showed that rVP2 exhibited poly (C)-dependent poly (G) polymerase activity, suggesting that the amino-terminal Histag did not affect the RNA polymerase activity of rVP2.
Partial characterization of rVP2 indicated that its RNA polymerase activity required the divalent cation Mg2+; the optimal concentration was 10 mmol/L. This was consistent with an early report of the properties of RNA transcriptase in reovirus subviral particles (Levin D H, et al., 1970), and it was also consistent with data on the VP1 protein of bluetongue virus, for which the optimal Mg2+ concentration for polymerase activity was found to be between 4 mmol/L and 8 mmol/L (Wehrfritz J M, et al., 2007). For the rotavirus RNA polymerases, optimal activity in the synthesis of dsRNA by rVP1 and rVP2 has been found to require unusually high concentrations of Mg2+, between 25 and 30 mmol/L (Boyce M, et al., 2004; Mansell E A, et al., 1990; Patton J T, et al., 1997). This suggests that the concentrations of Mg2+ that are critical for RNA polymerase activity vary between different viruses. With regard to temperature, we observed that the optimal temperature for the RdRp activity of rVP2 was 28 ℃, by comparison with the activity determined at 37 ℃ and 20 ℃ (Figure 4A), which is in agreement with the replication temperature for aquareoviruses. The optimal temperature for RNA synthesis in BTV, rotavirus and MRV, on the other hand, is 32 ℃ or 37 ℃ (Boyce M, et al., 2004; Patton J T, et al., 1997; Wehrfritz J M, et al., 2007). This suggests that the temperature for optimal RNA polymerase activity relates to the temperature at which replication occurs in host cells. It was notable that, in our study, the nucleotide incorporation rate observed for rVP2 was a little lower than that observed for GCRV particles, suggesting that the internal RNA polymerase (VP2 protein) located within the core shell is more stable than the 'unprotected' RNA polymerase of the recombinant VP2 in vitro. Lastly, we observed that the full-length rVP2 expressed in a baculovirus was active, whereas a truncated version, rVP2390-900, expressed in prokaryotic cells, was inactive. To characterize the catalytic activity of VP2 RdRp, it will be necessary for us in the future to analyze other parts or domains of VP2, as well as the central RdRp region, both in isolation and in association with each other.
To summarize, a recombinant version of the VP2 protein of GCRV was first constructed and successfully expressed in insect cells as a fusion protein with a Histag. The replicase activity of the purified rVP2 was assayed using poly (C) /oligo (G) as a template in vitro, in the absence of other viral proteins. This showed that rVP2 exhibited poly (C)-dependent poly (G) polymerase activity, and that this RNA polymerase activity required divalent Mg2+ cations and functioned at an optimal temperature of 28 ℃. This study represents the first characterization of the VP2 protein, and it provides a basis for further work to identify the site of RNA polymerase activity in GCRV VP2 by specific-site mutation or truncation analyses.
-
This work was supported by funding from the National Natural Science Foundation of China (grants: 31172434, 31372565).
-
All the authors declare that they have no competing interest. This article does not contain any studies with human or animal subjects performed by the any of the authors.
-
QF designed the experiments. LMY carried out the experiments. QF, LMY, HL and XML analyzed the data. LMY, QF, HL and XML wrote the paper. All authors read and approved the final manuscript.

 DownLoad:
DownLoad: