











-
Hepatitis C virus (HCV) constitutes a significant health burden worldwide, with an estimated 130-170 million people chronically infected. Severe liver disease, including advanced fibrosis, cirrhosis and hepatocellular carcinoma, is often a complication of long term infection, making HCV the most common indication for liver transplantation in developed countries (Thomas D L, 2013). Besides liver disease, persons with chronic HCV infection also have a high prevalence of multiple extrahepatic conditions compared to agematched, general population controls. Many of the extrahepatic outcomes of HCV infection are linked to the deregulation of B cells. A causative role of HCV in the generation of B cell lymphoproliferative diseases in HCV chronic carriers is strongly supported by numerous epidemiological and clinical observations and experimental data. The most common HCV associated B cell lymphoproliferations include type Ⅱ mixed cryglobulinemia (MC) and B cell non-Hodgkin lymphomas (B-NHL) (Agnello V, et al., 1992; Dammacco F, et al., 2000). Type Ⅱ MC is characterized by the monoclonal or oligoclonal proliferation of B cells in the bone marrow, liver tissue and peripheral blood (PB). These clonal B cells are responsible for the production of monoclonal IgM with anti-IgG rheumatoid factor activity. Circulating mixed cryglobulins are detected in 40%-60% of HCV-infected patients whereas overt cryglobulinemia vasculitis develops in only 5%-10% of the cases (Saadoun D, et al., 2007). This syndrome is due to the deposition of cryoglobulin-containing immune complexes on small and medium sized blood vessels causing inflammation in skin, kidney and/or other tissues (Agnello V, et al., 1992).Cutaneous manifestations are the most typical clinical signs of cryoglobulinemic vasculitis, represented mainly by cutaneous purpura of the lower limbs and chronic cutaneous ulcers typically located at the supramalleolar regions. The most frequently affected internal organs are the peripheral nerves, kidneys, and joints and the prognosis of the disease usually depends on the degree of the cryoglobulinemic damage of vital organs and on the presence of comorbidities (Ramos-Casals M, et al., 2012). Treatment of MC aims at the elimination of the causes of the disease, namely at the eradication of HCV, elimination of B cell clonal expansions and depletion of cryoglobulins. The triple therapeutic combination of pegylated-interferon-α plus ribavirin (a nucleoside antimetabolite agent) plus rituximab (a chimeric monoclonal antibody directed against the CD20 antigen) has been proven to be successful in 54% of HCV patients with cryoglobulinemic syndrome (Dammacco F, et al., 2010). Even though successful treatment of HCV infection results in the resolution of cryoglobulinemic vasculitis in the majority of cases, the use of the new direct-acting antivirals like Telaprevir or Boceprevir represents a further therapeutic option in MC patients who do not achieve a sustained virologic response or show a persistent cryoglobulin production despite virus eradication (Levine J W, et al., 2005).
HCV-associated B-NHL are of various subtypes, with splenic marginal zone B cell lymphomas and diffuse large B cell lymphomas being the most common among HCV-associated lymphomas. Indolent HCV-associated B-NHL can be treated with anti-viral therapy, whereas aggressive B-NHL in HCV patients iscurrently mostly treated with anthracycline-based chemotherapy and rituximab(Hermine O, et al., 2002; Peveling-Oberhag J, et al., 2013). Patients with B-NHL and chronic HCV infection have a worse clinical outcome compared with B-NHL patients without HCV-association, most likely due to the associated liver dysfunction (Peveling-Oberhag J, et al., 2013).
Two mechanisms have been proposed to explain the role of HCV in the development of B cell lymphoproliferative disorders, one involving chronic antigenic stimulation of B cells, the other -not mutually exclusive -a direct mutagenic effect of HCV on B cells (Figure 1). The regression of HCV-related B-NHL following antiviral therapy probably represents the strongest evidence of a link between HCV infection and the development of B cell lymphomas (Hermine O, et al., 2002). Based on this observation, chronic antigen stimulation has been suggested as a potential mechanism by which HCV drives B cell clonal expansions. This hypothesis is supported by the finding that HCV-associated lymphomas are derived from germinal center or post-germinal center B cells, indicating that lymphomagenesis occurs when B cells experience somatic hypermutation and proliferate in response to an antigen (De Re V, et al., 2000). Accordingly, the characterization of the immunoglobulin (Ig) heavy chain V region genes of the expanded clones in HCV-associated MC and B-NHL shows a restricted repertoire of V genes and the expression in HCV- positive NHL of a B cell receptor (BCR) with specificity towards viral antigens (Chan C H, et al., 2001; Quinn E R, et al., 2001). Clonal B cell populations, present in the liver and the PB of HCV-infected MC patients, demonstrate a preferential usage of the VH1-69 and Vκ3-20 gene segments, which are known to be used both by antibodies with rheumatoid factor activity and HCV E2 protein specificity (Charles E D, et al., 2008). The cloning and the expression as a soluble immunoglobulin of the BCR from a HCV- positive B cell lymphoma indeed revealed specificity of the BCR for the E2 protein, suggesting that the B cell lymphoproliferation might start from B cells which have been activated by the HCV envelope protein E2 (Carbonari M, et al., 2005; De Re V, et al., 2000; Ivanovski M, et al., 1998; Quinn E R, et al., 2001).
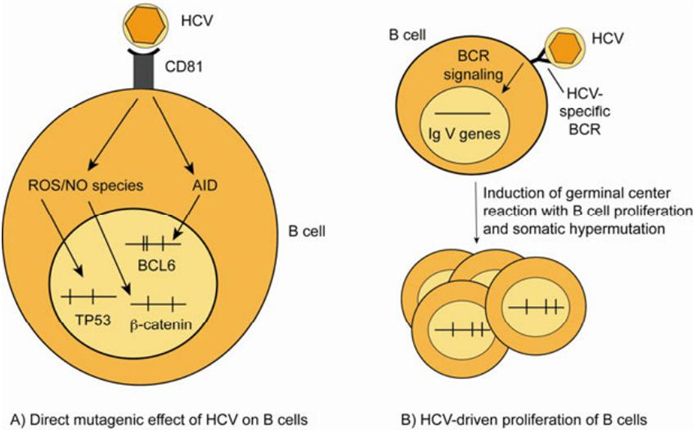
Figure 1. Scenarios for the role of HCV in the development of B cell lymphoproliferative disorders. (A) HCV induces mutations of cellular genes in B cells in vitro and in PB mononuclear cells from chronically HCV-infected patients. It was proposed that mutations in the genes TP53 and β-catenin are caused by reactive oxygen species induced upon binding of HCV to CD81 on B cells, and that increased mutation frequencies in BCL6 are due to HCV-induced upregulation of activation induced cytidine deaminase (Machida K, et al., 2004b; Machida K, et al., 2004a). (B) The chronic antigenic stimulation of B cells by HCV leads to a B cell proliferation that eventually may become monoclonal. B cells bearing a B cell receptor (BCR) with high affinity towards viral antigens may be selected in germinal center reactions, where they undergo somatic hypermutation (SHM). Apparently, this is accompanied by the development of B cell clones expressing BCR with (additional) rheumatoid factor activity. Vertical lines in the genes in A and B indicate somatic mutations. ROS: reactive oxygen species; NO: nitric oxide; AID: activation induced cytidine deaminase.
A second proposed mechanism how HCV might cause B cell lymphoproliferations, which is based mainly upon in vitro findings, involves a direct mutagenic effect of the virus on B cells. In particular, it has been reported that, upon exposure to HCV virions, B cell lines accumulate mutations in three target genes, the tumour suppressor gene TP53 and the protooncogenes CTNNB1 (β-catenin) and BCL6 (Machida K, et al., 2004a). One mechanism how HCV could be mutagenic is through the induction of a cellular oxidative stress state where some HCV proteins can induce the increase in the cellular levels of reactive oxygen species and nitric oxide, which leads to DNA damage (Machida K, et al., 2004a). In the in vitro studies TP53 and β-catenin were reported to be mutated with remarkably high mutation frequencies of 5-11×10-4/bp and proposed to be target genes of the reactive oxygen species (Machida K, et al., 2004b). A secondmutagenic effect of HCV is presumably mediated through upregulation of activation induced cytidine deaminase (AID) and error prone DNA polymerases ζ (zeta) and ι (iota) (Machida K, et al., 2004a). AID is the master regulator of somatic hypermutation of Ig V genes, and error-prone DNA polymerases are thought to contribute to the hypermutation mechanism. Somatic hypermutation physiologically targets also the BCL6 protooncogene at low frequency in the germinal center reaction, resulting in about a third of post-germinal center memory B cells carrying mutations in the major mutation cluster in intron 1 of BCL6 (Pasqualucci L, et al., 1998). In the in vitro studies, increased Ig V gene and BCL6 mutations were detected in the B cell lines (Machida K, et al., 2004a). Curiously, enhanced mutations were also reported in BCL6, TP53 and β-catenin in PB mononuclear cells from HCV-infected patients, although such isolates usually consist of only around 10% B cells (90% of PB mononuclear cells are T cells, monocytes and natural killer cells). In particular for BCL6, it is difficult to envision that this B cell-specific mutation target is broadly mutated in all types of blood cells.
It is important to mention that the in vitro studies on the mutagenic effect of HCV on B cells did not address the issue whether HCV has to enter the B cells to exert its effects. It is indeed a much debated and still unresolved question whether HCV infects B cells (Ito M, et al., 2010). The presence of HCV genomic RNA has been detected not only in serum and liver tissue but also in association with PB mononuclear cells of patients with chronic infection (Ferri C, et al., 1993; Inokuchi M, et al., 2009). B cells, monocytes, dendritic cells, and T cells have been reported to have detectable levels of HCV RNA or proteins (Bare P, et al., 2005; Fournillier A, et al., 2004; Zehender G, et al., 1997). However, although detection of the HCV negative strand RNA (the replicative intermediate) and viral proteins has been reported, there is no clear proof of a complete productive replication of HCV in B cells or in other extrahepatic compartments (Pham T N, et al., 2008; Sansonno D, et al., 2007). Distinguishing RNA association from true HCV replication is difficult, as the detection of HCV RNA within blood cells is not a proof for viral replication in these cells. Multiple artefacts complicate detection and quantitation of the replicative intermediate minus strand RNA (Lanford R E, et al., 1995; Lerat H, et al., 1998), mainly due to the lower concentration of the negative strand RNA as compared to the positive strand. The amount of the negative strand in the serum and liver is usually 10-100 times lower than the positive strand (Fong T L, et al., 1991). The issue of HCV infection of PB cells has also been addressed by using cell culture derived HCV, a robust in vitro system which recapitulates the full virus life cycle (Zhong J, et al., 2005). However, this approach demonstrated that immune cells, including B cells, do not support infection with cell culture derived HCV of genotype 2a (Marukian S, et al., 2008). Given the impact of HCV infection on the immune system, and the related extrahepatic manifestations, it is of crucial importance to clarify the mechanisms by which HCV RNA may associate with PB mononuclear cells and to reveal which types of haematopoietic cells support viral replication.
Importantly, it is possible that HCV can exert a mutagenic effect even without infecting and replicating in B cells. As HCV binds to the CD81 receptor on B cells, which is a B cell costimulatory molecule and builds complexes together with CD19 and CD21 (Levy S, et al., 2005), it can be envisioned that signalling through this receptor (and perhaps in addition through the BCR of HCV-specific B cells) could induce AID and perhaps also nitric oxide synthase in B cells, without the need to infect the B cells. In support of this, incubation of the HCV protein E2 with B cells is sufficient to cause upregulation of AID and DNA strand breaks in B cells (Machida K, et al., 2005). The exact mechanism how CD81 triggering induces reactive oxygen species and AID is still unclear. Notably, binding of HCV to CD81 on B cells may promote B cell lymphoproliferation not only by a mutagenic effect, but also by stimulating the B cells and promoting their survival, as binding of HCV particles to CD81 (without infection of B cells) causes NF-κb activation, upregulation of the anti-apoptotic factor BCL2, increased resistance against CD95-mediated apoptosis, triggering of the JNK pathway, and upregulation of the costimulatory factors CD80 and CD86 (Chen Z, et al., 2011; Levy S, et al., 2005; Rosa D, et al., 2005).
As most of the studies indicating a mutagenic effect of HCV on B cells were performed in vitro, we wondered whether such an effect might also be detectable in B cells in chronic HCV patients. Therefore, we analyzed the genes TP53, β-catenin and BCL6 for mutations in vivo, in B cells isolated from the PB and the liver of HCV chronic carriers (Tucci F A, et al., 2013). To assess whether virus-induced mutations occur in pre-or post-germinal center B cells, we studied the mutational status of TP53, β-catenin and BCL6 in naive and memory B cells from the PB of four chronically HCV infected individuals. Unexpectedly, the PB B cells did not harbour TP53 or β-catenin mutations in naive or memory B cells above the experimental background. Mutations were consistently detected in the BCL6 gene in PB memory B cells. However, BCL6 is a physiological target of somatic hypermutation, and the frequencies of BCL6 mutations identified in memory B cells of HCV patients were similar to those previously reported for memory B cells in healthy humans (Pasqualucci L, et al., 1998; Seifert M, et al., 2009). No BCL6 mutations were found in naive B cells. Therefore, we conclude that the BCL6 gene is not undergoing increased mutation accumulation in PB B cells of HCV- positive individuals. In line with our observations, a recent study of MC and HCV-associated B cell lymphomas did not detect increased BCL6 mutation levels in these lymphoproliferations of HCV patients (Hofmann W P, et al., 2007).
HCV replication is primarily taking place in the liver, and the presence of intrahepatic lymphoid aggregates is a typical feature of chronic HCV infection. These aggregates resemble germinal centers, which are characterized by oligoclonal populations of strongly proliferating B cells (Racanelli V, et al., 2001; Sansonno D, et al., 2004). In the intrahepatic B cell aggregates, the interaction between HCV and B cells may be more direct as compared to the PB. In our analysis of CD20+ B cell aggregates microdissected from the livers of two HCV- positive patients, however, no mutations in TP53 and β-catenin were identified (Tucci F A, et al., 2013). The mutation frequency determined for the BCL6 gene was even lower than the one typical of PB memory B cells. This is likely due to the fact that the intrahepatic follicles are composed of a mixed population of pre- and post-germinal center B cells (unpublished own observation). Additionally, some non-B cells with unmutated BCL6 gene were likely present among the isolated cell clusters.
In conclusion, based on the findings from the PB and the liver of six HCV-infected individuals, it emerges that B cells in these patients do not acquire HCV-driven mutations in the three genes analyzed. Hence, further work is needed to clarify the role of HCV in the pathogenesis of B cell lymphoproliferative disorders in chronic virus carriers, and whether HCV's contribution to the development of these diseases includes direct mutagenic effects. As methods are now available to study somatic mutations genome-wide by next generation sequencing, it will be interesting to perform such studies for HCV-associated B cell lymphoproliferations to find out whether such clonal B cell expansions have a specific mutation pattern as a genetic trait of the mutagenic effect of HCV.
HTML
-
This work was supported by the DFG through SFB TRR60.

 DownLoad:
DownLoad: