









HTML
-
Influenza A viruses are among the most devastating diseases from both a public health and an economic viewpoint. Thousands die annually from influenza A virus infection. Moreover, this virus causes millions of deaths in different species of commercial birds (Nili H, et al., 2003). Therefore, more attention is focused on type A influenza viruses, primarily the possible pandemic potential of these infectious agents. Public health officials in many countries are especially concerned about the emergence of new subtypes of influenza A viruses. Owing to the high mutation rate in the genomic segments encoding the influenza hemagglutinin (HA) antigen, conventional vaccines are unable to protect against the emergence of new viruses (Cox N J, et al., 2000). Each year, influenza vaccines must be updated to follow drift and shift virus changes. One of the new approaches to vaccine development is the targeting of conserved antigens in order to generate cross-strain immune responses and protection, which is becoming vital (Fu T M, et al., 2009).
M2, the third integral membrane protein, is a highly conserved influenza protein and is viewed as a potential universal vaccine target. M2 protein functions as a tetrameric proton channel; this protein hardly exists on the virus particles but is frequently seen on cells infected with the virus and possesses an important function in uncoating viral particles in endosomes for viral entry and maturation of HA during viral exit (Holsinger L J, et al., 1991; Lamb R A, et al., 1985; Pinto L H, et al., 1992). The N-terminus of M2 (M2e) is a highly conserved site throughout all influenza A viruses and has been reported to be a good epitope in controlling virus infection (Babapoor S, et al., 2012; Fiers W, et al., 2001; Fiers W, et al., 2004; Liu W, et al., 2005; Macken C, et al., 2001; Neirynck S, et al., 1999). M2e is only a 23 amino acid peptide, probably owing to its small dimensions and its position adjacent to several massive glycoproteins and glycolipids, which makes it difficult for the immune system to recognize it (Black R A, et al., 1993; Swinkels WJ, et al., 2013); however, it is possible to increase the size and antigenicity of this protein to facilitate recognition by the immune system and lead to a much stronger immune response. This can be achieved by linking the peptide to an appropriate carrier and, in several studies, it was found that the antibodies and cellular immunity effectively protected mice against influenza virus infection. Two basic methods—chemical conjugation and genetic insertion—have been used for this purpose. The molecules most commonly used for chemical conjugation are: keyhole limpet hemocyanin (KLH), Neisseria meningitidis outer membrane protein complex (OMPC), bovine serum albumin, glutathione transferase, a synthetic multiple antigen peptide, flagellin, and cholera-toxin-derived CTA1-DD (Ernst W A, et al., 2006; Fan J, et al., 2004; Frace A M, et al., 1999; Huleatt J W, et al., 2008; Liu W, et al., 2003; Liu W, et al., 2004; Mozdzanowska K, et al., 2005; Mozdzanowska K, et al., 2003; Tompkins S M, et al., 2007). In the second most frequently used strategy, the following insertion molecules are used: tetrameric domain of GCN4 (general control nondepressible 4), toll-like receptor 5 (TLR5) ligand, Salmonella typhimurium flagellin type 2 (STF2) and hepatitis B virus core (HBc) particle (Bessa J, et al., 2008; De Filette M, et al., 2008; De Filette M, et al., 2008; Ebrahimi S M, et al., 2012; Ebrahimi S M, et al., 2010; Huleatt J W, et al., 2008; Ionescu R M, et al., 2006; Mirzaei N, et al., 2014; Neirynck S, et al., 1999).
The conserved heat shock protein family, commonly used for genetic insertion, is widely distributed throughout microorganisms and mammalian cells. Heat shock proteins (Hsps) are amongst molecules that are able to function as the second signal, stimulate antigen-presenting cells (APCs) to secrete proinflammatory cytokines (TNF-α and IL-6), and strongly promote Th1 responses (Segal B H, et al., 2006). Moreover, Hsps can induce macrophages to produce nitric oxide and increase the production of co-stimulatory molecules that are needed for priming naive T cells. Thus, Hsps possess an independent immunostimulatory capacity which has made them attractive reagents for the induction of specific immune reactions.
In recent years, Hsps and molecular chaperones have also been suggested as genetic adjuvants for the induction of immunologic responses (Chen C H, et al., 2000; Qazi K R, et al., 2005). Mycobacterial has an antigenic nature which enables it to be used as a vaccine candidate against mycobacterium; furthermore, this property facilitates its application as a potential immune carrier molecule under certain circumstances. Li et al. stated that only the terminal portion of Hsp70 functions as an adjuvant (Li X, et al., 2006).
Previous studies have shown, the M2e protein of influenza A virus, when is genetically fused to the N-terminus of the truncated Hsp70 molecule of Mycobacterium tuberculosis (Hsp70359-610) and expressed in E. coli (Ebrahimi S M, et al., 2010) and Pichia pastoris (Ebrahimi S M, et al., 2010), can greatly enhance the immunogenicity of the M2e protein (Ebrahimi S M, et al., 2011; Jazi M H, et al., 2012).
Thus, in the current study, we designed the M2e. Hsp70c recombinant protein as a candidate for universal influenza A vaccine, tested in the C57BL/6 mouse model, and found mice vaccinated with M2e.Hsp70c-based vaccines were protected against challenge with influenza viruses.
-
Cloning, expression and purification of M2e.Hsp70c constructs were performed as previously described (Ebrahimi S M, et al., 2010).
-
One hundred and thirty 6-week-old female BALB/c mice were obtained from the Animal Rearing Department of the Pasteur Institute, Iran. All of the mice were randomly divided into ten equal groups of 13 at 7 weeks old. The animals were maintained in a controlled-temperature environment with 12h-light/12h-dark cycles and given food and water ad libitum. Mice were kept for 1 week in the animal room for adaption and immunized when they reached 8 weeks of age (Table 1).
Group Vaccination Challenge virus Total no. of mice 1 M2e-Hsp70 PR/8(H1N1) 13 2 M2e PR/8(H1N1) 13 3 Hsp70 PR/8(H1N1) 13 4 Split vaccine PR/8(H1N1) 13 5 No(PBS) PR/8(H1N1) 13 6 M2e-Hsp70 H9N2 13 7 M2e H9N2 13 8 Hsp70 H9N2 13 9 Split vaccine H9N2 13 10 No(PBS) H9N2 13 Table 1. Experimental design, vaccination status and number of mice in control and treatment groups.
All experiments were performed in accordance with conditions stated by law and authorized by the Institutional Ethical Committee on Experimental Animals, Razi Vaccine and Serum Research Institute of Iran.
-
Iranian isolates of influenza viruses H1N1 were a kind gift from Dr Mokhtari (Virus Research Dept, Tehran University of Medical Science, Tehran, Iran), and propagated in Madin-Darby canine kidney (MDCK) cells. Avian influenza virus, H9N2, was a kind gift from the Marand branch of Iran's Razi Vaccine and Serum Research Institute, and passaged in allantoic fluid of 10-day-old specific-pathogen-free (SPF) embryonated chicken eggs.
-
Experimental mice were immunized at the age of 8 weeks and boosted 2 weeks later by the (s.c.) route without endotoxin. As shown in Table 1, five groups were allocated for experimental study of each of the H1N1 and H9N2 influenza A viruses. In the treatment groups, 30 μg of M2e-Hsp70c, M2e peptide and Hsp70c were used for immunization in groups one to three, respectively; the fourth group was used for split vaccine and the fifth for negative control (only buffer).
-
In order to synthesize the M2e peptide (23 amino acids, 2-24, SLLTEVETPIRNEWGCRCNDSSD) based on the consensus sequence of A/Chicken/Iran/101/1998 (H9N2) influenza virus, solid-phase technology (GL Biochem, China) with a minimum purity of 95% was used. Subsequently, the peptide was employed in the anti-M2-specific ELISA (enzyme-linked immunosorbent assay).
-
Blood samples were taken from a ventral tail vein in all groups before—and 2 weeks after—immunization. Samples were kept at 37℃ for 60 min and then placed on ice; serum was separated after two successive centrifugations.
The ELISA was performed with some modification as described previously (Zhao G, et al., 2010). Briefly, 96-well plates (MaxiSorp, Nunc, Denmark) were coated with 100 µL of 10 μg/mL M2e synthetic peptide solution in 50 mmol/L sodium bicarbonate buffer, pH 9.6, and incubated overnight at 4℃. The plates were then washed with washing buffer and blocked for 1 h with PBS-BSA 2% at room temperature. The plates were loaded with 1/200 dilution of experimental sera and incubated for 2 h at 37℃ and, after washing, 100 µL of 1:10000 rabbit anti mouse IgG-HRP conjugates (Sigma-Aldrich, St. Louis, USA) were added to the wells and incubated for 2 h at 37℃. The color reaction was developed with 3, 3', 5, 5'-tetramethylbenzidine, TMB (Pishtaz Teb, Tehran, Iran) at optical density (OD) 450 nm.
The ODs of the experimental groups at 1/200 dilution were compared with each other and with those of the control groups.
-
Interferon γ(IFN-γ) ELISPOT (enzyme-linked immunospot) assay was performed as described (Tompkins S M, et al., 2007) on mouse spleen cells by stimulation with M2e peptides. A total of 2×105 spleen cells were placed in each well of a 96-well plate and in vitro stimulation carried out with 10 μg/mL of M2e peptide, as positive control cells stimulated with PHA (phytohemaglutinin), and wells containing un-stimulated cells and RPMI-1640 (Gibco®, USA) were used as negative controls. The number of cells secreting M2e-specific IFN-γ in the mice were counted using commercial ELISPOT assay kits (Mabtech AB, Sweden). This procedure was conducted according to the instruction manual. The visible spots of IFN-γ-secreting cells were enumerated using Cellular Technology Ltd (USA) software.
-
To detect the cytotoxicity of the candidate vaccine, the carboxyfluorescein succinimidyl ester (CFSE, Invitrogen, Carlsbad, CA, USA) method was used in this study (Bijker M S, et al., 2007). Splenocytes from naive mice were stained with 5 μmol/L (high intensity) or 0.5 μmol/L (low intensity) of CFSE. The high-intensity CFSE-labeled cells were loaded with M2e peptide 10 μg/mL while the low-intensity CFSE-stained cells (0.5 μmol/L) were used as a peptide un-pulsed control. The cells were then transferred intravenously (3×106 cells of each) into the experimental groups of mice. After 20 h, the lymphocytes were isolated from spleen and resuspended, and RBCs were lysed and flowcytometry analysis was then carried out to highlight the difference in separation pattern based on intensity of CFSE staining. The percent killing of the high-and low-intensity CFSE-labeled cells was calculated and the specific killing of M2e pulsed (CFSE high) target cells was calculated as follows:(1-((CFSE high/CFSE low) vaccinated ×(CFSE low/CFSE high) naive))× 100%.
-
At 2 weeks after the last immunization, the mice were challenged intranasally (i.n.) with a lethal dose (1×107 pfu) of A/PR8 and H9N2 in a total volume of 50 μL under mild anesthesia with ketamin/xylasine solution.
Three mice were selected randomly in each group to represent lung viral titration on days 3, 6 and 9 post challenge, and for 2 weeks other mice were monitored daily in each group for mortality and morbidity following monitoring of body weight and observation of clinical symptoms at 2-day intervals.
-
To verify the impact of M2e-based vaccines on viral replication in the respiratory system of mice, viral shedding was evaluated by taking a throat swab from the mice on different days after challenge. Three mice from each group on days 3, 6 and 9 died following challenge via cervical dislocation under deep anesthesia with ketamin/xylasine.
After removal, the lungs were aseptically homogenized for evaluation of viral load. Next, the lungs were titrated by inoculation of decimal dilutions in Madin-Darby canine kidney (MDCK) cell cultures, as described previously (Rimmelzwaan G F, et al., 1998).
Viral titers of the lung homogenates are shown as log10 geometric mean titer of TCID50/mL 7SEM, and infectious titers were established according to the method described by Reed and Muench (Reed L J, et al., 1983) using BPL-treated Iran/H1N1, or Iran/H9N2 antigens.
All negative samples for the virus titration test were set at 0.9 log10 TCID50/mL.
The lungs of infected mice were fixed in 10% neutralbuffered formalin for histopathologic examination on day 7 after challenge for evidence of cellular inflammation and necrosis.
Five-micrometer paraffin-embedded sections were used for histopathologic examination following staining with hematoxylin and eosin (H & E).
-
SPSS 16.0 software (SPSS Inc., Chicago, IL, USA) was used to statistically compare the results of the treatment and control groups. Data were analyzed for significance (P ≤ 0.05) via one-way ANOVA (analysis of variance) when the variance between groups was homogeneous and distribution of the data was normal, and when the normality test or homogeneity of variance test was unsuccessful, a nonparametric (Kruskal-Wallis) test was used.
Cloning, expression and purification of M2e. Hsp70c (Hsp70359-610)
Mice
Virus preparation and titration
Immunization
Peptides
ELISA
ELISPOT
CTL assay
Flowcytometry
Viral challenge and follow-up
Viral shedding and histopathologic examination
Statistical analysis
-
Total IgG (immunoglobulin-gamma) was evaluated with an indirect ELISA method. The results showed that immunization of the mice with prokaryotic M2e-Hsp70c induced specific IgG antibody responses that showed significant differences compared with those of the control groups (P ≤ 0.05). No significant differences were observed among the control groups (P > 0.05). These results are shown in Figure 1.

Figure 1. Humoral immune responses in C57/BLC. Anti-sera diluted to 1/200 was gathered from individual mice 2 weeks after the *first immunization (2WFI) and 2 weeks after second immunization (2WSI), separately. The bars represent average antibody responses for each group of immunized mice with standard error (mean ± S.E.M). Means with different letters between bars are statistically different at the same time point of immunization, 2WSI (P < 0.05, one-way ANOVA).
-
Strong positive spots were observed only after M2e peptide stimulation, while in the negative control group very few spots were observed (Figure 2). These signifi-cant differences—between the results of the control and treatment groups—emphasize the role of M2e peptide in the stimulation of M2e-specific T-cell response in the M2e.Hsp70c group.
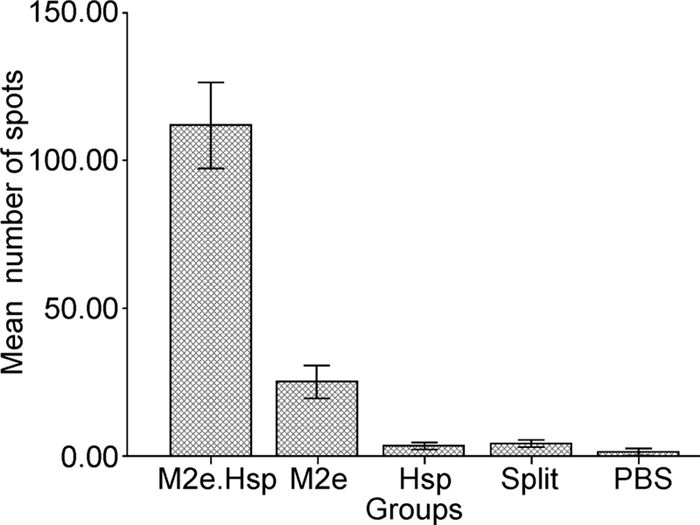
Figure 2. Immunization of mice with M2e.Hsp70 (Hsp70359-610) can activate M2e-specific T cells. In the mice immunized with these vaccines, lymphocytes were measured by ELISPOT in the presence of 5 μg/mL M2e peptide. Lymphocytes from un-immunized mice were used as controls, and the number of IFN-γ-secreting lymphocytes was counted in 2×105 spleen cells. The average for each group of immunized mice is shown with a bar with standard error (mean ± S.E.M). Means with different letters between bars are statistically different at the same time point of immunization.
-
For detection of cytotoxicity of the lymphocytes, an in-vivo CTL assay using the CFSE method was employed. The results showed that immunization of the mice with M2e-Hsp70 (Hsp70359-610) induced CTL activity compared with the control groups (P ≤ 0.05) (Figure 3). No significant differences were observed among the control groups (P > 0.05).

Figure 3. The ratio of CFSE low/CFSE high cells was determined by flow cytometry by gating on CD45.1+ lymphocytes. Specific killing of M2e pulsed (CFSE high) target cells was calculated as follows: (1-((CFSE high/CFSE low) vaccinated × (CFSE low/CFSE high) naive))×100%. A1, PBS (negative control); A2, M2e; A3, Hsp70; A4, split vaccine; A5, M2e.Hsp70c.
-
Following challenge, the mice were monitored for weight changes and mortality every 2 days (Figure 4). As expected, a lethal dose challenge (1×107 pfu) of A/PR8 and H9N2 viruses caused 80% mortality in the non-vaccinated groups; however, 100% of mice vaccinated with M2e-Hsp70c survived against both H1N1 and H9N2 viral challenges and after 8 days post challenge began to recover.
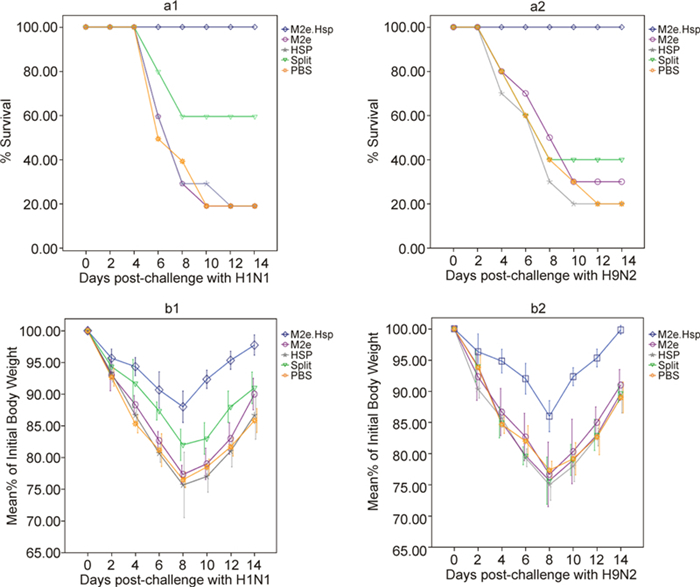
Figure 4. Results of prime-boost immunization with recombinant fusion proteins administered to mice (10 mice/group) and challenges with H1N1or H9N2 influenza A isolates. Mice immunized with PBS were challenged as negative controls. Survival rate (a1-a2: panels are based on viral challenge isolate) was monitored on a daily basis; percent of initial body weight (b1-b2: panels are based on viral challenge isolate with standard error bars) was monitored at 2-day intervals for 2 weeks after viral challenge.
Of the mice vaccinated with split vaccine, 60% survived after challenge with H1N1 and just 40% survived after challenge with H9N2. Therefore, compared with the commercial vaccine (split vaccine), M2e-based vaccine protected mice against heterologous virus challenge. Basically, the differences in survival rate in the injected group compared with the PBS group or split control groups were highly significant (P ≤ 0.05). All mice in the challenged groups showed weight loss. In non-immunized mice and mice immunized with the split vaccine, weight loss was more significant.
The results of this study clearly show that mice vaccinated with M2e.Hsp70c-based vaccines are protected against challenge with influenza viruses.
-
All mice in the un-challenged negative control group did not shed any virus, were negative throughout the study period, and showed no adverse clinical signs.
Three and four days after H1N1 and H9N2 challenge respectively, the unvaccinated challenged group showed evident clinical signs, including ruffled fur, hunched posture, rapid breathing, inactivity and paralysis of posterior limb.
The clinical signs in the mice vaccinated with the M2e-based vaccines, and challenged, appeared with a delay of 2 to 3 days. Mild symptoms were observed for 3-4 days, and then the mice recovered after 1 week.
Mice immunized with the split vaccine, challenged with heterologous virus H9N2, developed more clinical symptoms compared with mice challenged with the homologous virus, H1N1.
Gross examination of the lungs during the necropsy of the mice in the unvaccinated challenge group and the mice vaccinated with split vaccine at 6 days post infection revealed multiple foci of hemorrhage on the lungs surface. However, mice immunized with the M2e-based vaccines revealed few focal hemorrhages.
Microscopic observation of the lungs showed bronchial epithelial necrosis, congestion, edema and lymphocytic infiltration in all challenged groups.
Comparative analysis of serum antibody responses against M2e peptide using the ELISA technique
ELISPOT results
CTL results
Challenge results
Clinical signs, histopathologic examination and viral shedding
-
Influenza A outbreaks in the human and animal population are a concern for both human public health and veterinary officials. Between 500 and one million deaths are caused by influenza epidemics annually worldwide, and such epidemics are also a huge economic burden (WHO, 2002). The global pandemic that will eventually come shall bring at least 10 to 50 times more destruction.
An ideal way to deal with this threat is a vaccine that protects efficiently against all influenza A strains, provides long-term immunity, and is affordable to produce. Using vaccines that have cross-protection between different strains will reduce the costs of production and will ultimately increase their availability for public health purposes.
Although one of the most troubling features of influenza A viruses is antigenic shift and drift on surface antigens, until now, the M2e sequence has not changed considerably from the first-known sequence (Reid A H, et al., 2002). This unique feature of M2e has attracted the attention of scientists and researchers in the field of vaccine research and development, since this protein can be used as a potential candidate for vaccine production against heterologous viruses.
Cross-reactive and protective antibodies are extracted by the highly conserved N-terminal sequence of M2e (residues 2-9)(Grandea A G, et al., 2010).
It is clear that M2e is not subjected to selection pressure for drift or shift antigenic change. However, one of the main problems with M2e is its weak immunogenicity. Because this is a small peptide close to the cell membranes and in its native form is a very weak immunogen, only a small portion of infected individuals produce anti-M2e antibodies (Black R A, et al., 1993) and there is no indication that M2e low-titer antibodies play an important role in the protection against new infections by heterologous strains.
Current approaches used to increase the immunogenicity of this antigen are based on several important strategies.
One of the most efficient approaches in this regard is using an appropriate vector to improve the effectiveness of transcriptional components using a Kozak-modified sequence to boost protein expression (Kutzler M A, et al., 2008). Toll-like receptor agonists such as Hsp70 will shift immune responses to cellular immunity.
Cellular immunity is very important in the control of viral infections and, therefore, by enhancing this function, viral infections can be inhibited more effectively.
For example, the C-terminal domain of Hsp70359-610 (Hsp70c) is considered a strong regulatory molecule in the immune system that targets Toll-receptor 4 ligands.This terminal region of Hsp70 molecules is known to increase immunogenic APC function, creating strong CTL responses and preventing tolerance induction. It is also vital to induce polarized cytokine responses toward T helper 1 (Th1) cells, triggering the production of IgG2a antibody in mice and humans, which has an important function in protection (Bolhassani A, et al., 2008; Li X, et al., 2006). Nevertheless, immunization procedures and methods of delivery are essential in determining the signature and protection mechanism of cytokine patterns (Grandea A G, 3rd, et al., 2010; Wang S, et al., 2008).
Laboratory mice have proven to be a useful tool in measuring infectivity and pathogenesis, and, subsequently, in the evaluation of vaccine and antiviral candidates (Haga T, et al., 2010; Lu X, et al., 1999).
Assessment of the serological results determined that Hsp70359-610 Hsp70c) connected to M2e increased the anti-M2e antibody responses. The total antibody results show that using Hsp70c in this vaccine causes an increase in immune responses. Humoral immunity and total antibody play a key role in controlling viral infections. In fact, antibody will fuse to viruses and neutralize those that inhibit virus infection and outbreak of disease.(Ebrahimi S M, et al., 2012; Jazi M H, et al., 2012; Swinkels W J, et al., 2013).
However, IFN-γ significantly increased in ELISPOT results. IFN-γ has a critical role in the induction of cellular immunity responses. This cytokine, by stimulation of T CD8 (CD8 T cells), will destroy infected cells. The results show that the fusion of Hsp70 with M2e causes an increase in IFN-γ and that this cytokine has a key role in controlling influenza infection.
The results also show that M2e.Hsp70c significantly increases CTL activity. Control of viral infection depends on the action of T CD8. In fact, T CD8, by killing infected cells, will destroy virus rescores. Furthermore, these results show that the fusion of Hsp70 with M2e potentially induces CTL responses, which play an effective role in the control of influenza infection.
Finally, the challenge results showed a high survival rate in the M2e.Hsp70c group, which means better control of viral infection.
Viral infection control depends on both humoral and cellular immunity because Ab neutralizes viruses, and cellular immunity—by killing infected cells—destroys virus rescores, and by cooperation of both humoral and cellular immunity enables better control in viral infections. Our results confirmed cooperation of both humoral and cellular immunity in the M2e.Hsp70c group; however, the M2e group did not perform as well as the M2e. Hsp70c group in inducing humoral and cellular immunity and was unable to control viral infection.
It has been shown that antibodies and T cells play a vital role in protection against influenza virus (De Filette M, et al., 2005).
Delay in the appearance of clinical signs in vaccinated challenged mice for 2-3 days compared with unvaccinated mice shows the effectiveness of M2e-based vaccines in reducing the severity of the disease. Histopathologic analysis of the lungs of infected mice immunized with the fusion protein M2e.Hsp70c showed reduced tissue damage such as the accumulation of mononuclear cells, hemorrhage and pulmonary edema. This can be explained by the results of the present study in which influenza-virus-induced antibody was detected by day 7 after challenge and by previous studies in which systemic antibodies induced by influenza virus were first detected in the serum at day 5-6 after challenge (Carrat F, et al., 2008; Ebrahimi S M, et al., 2012; Jazi M H, et al., 2012; Lau L L, et al., 2010; Sealy R, et al., 2003). Immunization of mice with split vaccines did not lead to a reduced level of lung viral load, morbidity, mortality and histopathologic damage in lung tissues against lethal doses of viral challenge compared with mice immunized with M2e.Hsp70c. Although split vaccines induced a high level of HA-antibody in mouse, these derived antibodies are not effective in reducing the level and duration of viral shedding. The conventional split vaccine failure against H1N1and H9N2 isolates circulating in Iran can be explained by point mutation (antigenic drift) occurring in hemagglutinin (HA) (Seo S H, et al., 2002). Challenge failure with H9N2 avian influenza against split vaccine in this experiment was expected, since HA antigen in the split vaccine was used and the challenged virus was heterologous.
Dissimilar results and conclusions compared with previous studies may be due to the difference in carrier proteins, adjuvant or routes of administration in the induction of immune responses (Babapoor S, et al., 2012; Ebrahimi S M, et al., 2012; Jazi M H, et al., 2012).
In conclusion, the M2e.Hsp70c vaccine grants full protection to mice against a potentially lethal heterologous influenza virus infection. Developing the M2e antigen can overcome one of the main obstacles of vaccine production against influenza A viruses. Since this is a conserved protein among all human strains it could therefore provide good protection against heterologous subtypes. Further research is required on the mechanism of protective immunity, and on the efficacy of M2e.Hsp70c as an immunogen in new species. The fact that the M2e. Hsp70c vaccine increases protective antibodies for the purpose of protection leads us to conclude that this could be a "universal" influenza A vaccine.
-
The authors wish to thank Dr. Mehran Dabaghian and Dr. Mohammad Hossein Zabeh Jazi (faculty members of Razi Vaccine and Serum Research Institute), Dr. Mokhtari (Virus Research Dept, Tehran University of Medical Science), Dr. Seyyed Mahmood Ebrahimi (Applied Biotechnology Research Center, Baqiyatallah University of Medical Sciences) and Dr. Bin Gao (CAS Key Laboratory of Pathogenic Microbiology and Immunology, Chinese Academy of Sciences) for their helpful contribution to this project.
-
All the authors declare that they have no competing interest. All experiments were performed in accordance with conditions stated by law and authorized by the Institutional Ethical Committee on Experimental Animals, Razi Vaccine and Serum Research Institute of Iran.
-
Hamidreza Attaran conceived and coordinated the study, performed all experiments, and wrote the manuscript. Hassan Nili participated in the experiment design and manuscript drafting. Majid Tebianian carried out the immunoassays. All authors read and approved the final manuscript.

 DownLoad:
DownLoad: