









HTML
-
Viruses are intracellular parasites that depend on cellular components to replicate. Viruses communicate with the cellular machinery through protein–protein interactions (PPIs), for which each individual virus encodes a unique array of proteins. Each viral protein is capable of independently interacting with multiple cellular partners with various functions and, as a result, PPIs between viruses and cells have diverse roles that can provide everything required for viral replication, from the initial stage of viral entry through to the final stage of virus maturation. In addition to PPIs between viruses and cells, viral proteins also employ such interactions to assemble themselves into functional viruses. Homologous and heterologous interactions are two major forms of PPIs that frequently occur among viral proteins in the context of infected cells. These interactions usually lead to the formation of high-order oligomeric structures that are vital for the proper assembly of infectious and mature virions with the ability to spread infection within and between hosts.
In 2005, Barrios-Rodiles and colleagues developed a luminescence-based mammalian interactome mapping (LUMIER) method for detecting dynamic PPI networks in mammalian cells (Barrios-Rodiles M, et al., 2005). This methodology has been successfully used to map some signaling pathways (Braun P, et al., 2009; Miller B W, et al., 2009). LUMIER is similar in principle to the traditional coimmunoprecipitation (Co-IP) method, which allows detection of binary PPIs in a native condition. It involves the generation of two fusion constructs: one is an epitope-tagged protein (the bait protein) expression plasmid, while the other consists of a cDNA-expressing fused protein of the Renilla luciferase (RL) enzyme and a protein of interest (the prey protein). When coexpressed in mammalian cells (Figure 1A), their potential interactions are determined by performing an RL enzyme assay on the precipitated complexes that are precipitated by an antibody against the epitope tag.
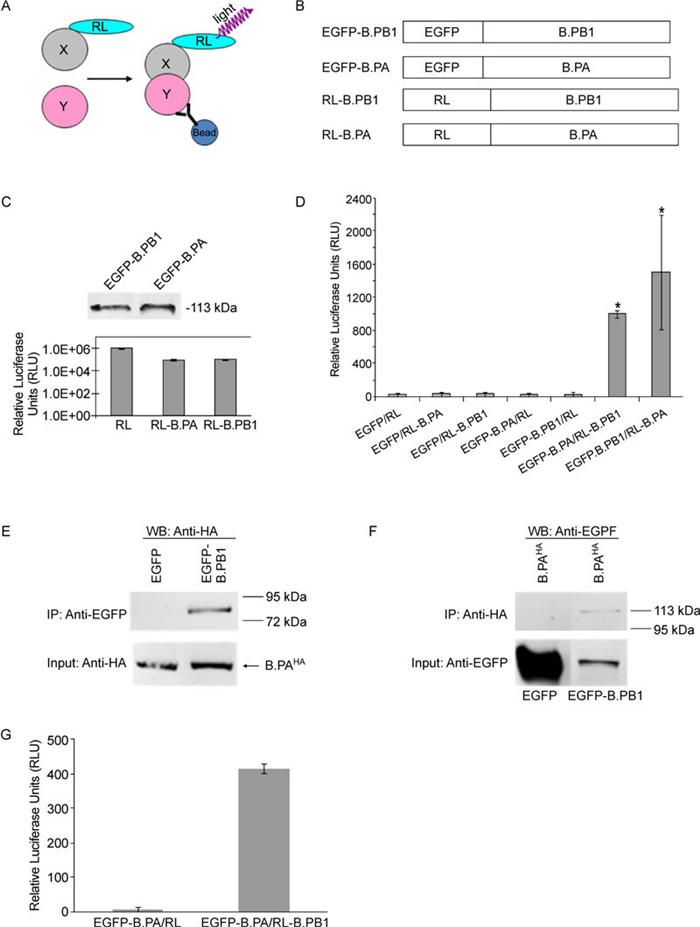
Figure 1. LUMIER-based strategy for the detection of PPIs, a Co-IP experiment, and the 96-well–format LUMIER assay. A: Schematic representation of the LUMIER principle for the detection of PPIs. The presence of a binary X–Y protein complex is determined by Co-IP using an anti-Y protein antibody followed by the detection of X-protein–associated luciferase activity on immunoprecipitates (Barrios-Rodiles M, et al., 2005; Braun P, et al., 2009; Miller B W, et al., 2009). B: Schematic representation of constructs used in the LUMIER system. C: Fusion protein expression profiles. For EGFP-derived constructs, levels of intracellular protein expression were determined by western blotting with an anti-EGFP antibody (Abcam). The number on the right indicates the migration position of molecular-mass markers (in kilodaltons). For RL and its derivatives, the levels of intracellular protein expression were confirmed by measuring luciferase activity on cellular lysates derived from transfected cells with each of the indicated plasmids (0.5 µg each). D: LUMIER detection of PPIs. COS-1 cells in a 12-well plate were transfected with RL or RL-tagged polymerase subunit constructs as indicated, with either EGFP or GFP-tagged constructs (0.5 µg of each plasmid). PPIs were determined by measuring RL activity on immunoprecipitates prepared using an anti-EGFP antibody. PPIs exhibiting a twofold or higher increase over background signals in the luciferase activity assay were empirically scored as positive (indicated by an asterisk). The data shown are a representative of three independent experiments. Each experiment was run in triplicate. E and F: Co-IP experiment. COS-1 cells grown in 60-mm Petri dishes were transfected with the indicated pairs of plasmids (3 µg each): B.PAHA with EGFP and B.PAHA with EGFP–B.PB1. Transfected cells were (E) first immunoprecipitated with anti-EGFP antibody, followed by probing with anti-HA antibody in western blotting (WB); or (F) first immunoprecipitated with anti-HA antibody, followed by probing with anti-EGFP antibody in western blotting. In brief, at 48 h post-transfection, the cells were lysed in RIPA buffer (150 nmol/L NaCl, 50 mmol/L Tris pH 8.0, 0.4 mmol/L EDTA, 10% glycerol, 1% NP-40) containing phosphatase and protease inhibitors. After brief centrifugation, cellular lysate samples were immunoprecipitated with either anti-GFP rabbit polyclonal antibody (panel A) (1:1000 dilution in a 5 mL reaction volume) (Abcam) or anti-HA mouse monoclonal antibody (panel B) (1:2000 dilution in a 5 mL reaction volume) (Sigma-Aldrich) and then incubated with 50 µL protein G Sepharose beads (Pierce). After extensive washing, the immunoprecipitates were analyzed in the Odyssey western blotting assay using appropriate combinations of primary and secondary antibodies. In (F) (i.e., western blotting with anti-EGFP), images in the left lane (i.e., EGFP) have been artificially placed together with those in the right lane (i.e., EGFP–B.PB1) for simplification, despite different molecular weights. The data were derived from same experiment. For the LUMIER assay in the 96-well format (G), COS-1 cells (approximately 5, 000 cells/well) in a 96-well cell culture plate were transfected with the indicated pairs of constructs (0.1 µg of each plasmid). At 48 h post-transfection, cells were lysed with Promega's luciferase kit lysis buffer (150 µL) on ice for 15 min with gentle shaking, followed by the addition of 0.5 µL anti-EGFP antibody (Abcam) to a 100 µL final volume. After 4 h shaking incubation at 4 ℃, 10 µL of protein A/G magnetic beads (Pierce) was added for an additional 4 h shaking incubation. After vigorous washing (six times) in pre-cooled PBS buffer, 20 µL of cell lysates together with 20 µL of PBS buffer, including magnetic beads, was transferred to a white 96-well plate (Pierce) and luciferase activity was then measured with a luciferase assay kit (Promega). The data shown are representative of three independent experiments. Each experiment was performed in triplicate, and the RLU value is the average of three wells.
We applied this technology to detect viral PPIs using a well-characterized interacting pair of influenza B polymerase proteins, PA and PB1. The experiments showed that specific PA–PB1 interactions were quantitatively detected on both 12-and 96-well–format LUMIER assays. Thus, our data support the utility of this assay in a high-throughput format for the study of viral PPIs.
Two groups of constructs were generated for the LUMIER assay (Figure 1B). One group involved the fusion of RL to the influenza B polymerase proteins (PA and PB1) at the N-terminus via a protein linker (GGGS×2), designated RL–B.PA and RL–B.PB1, respectively (Figure 1B). The other group was constructed by inserting an enhanced green fluorescent protein (EGFP) tag into the N-terminus of the same polymerase proteins, resulting in two fusion constructs designated EGFP–B.PA and EGFP–B.PB1. We used previously described epitope-based tagging strategies to detect polymerase subunits for this study, as antibodies capable of recognizing native proteins were not available (Deng T, et al., 2005; Deng T, et al., 2006; Fodor E, et al., 2004; Gonzalez S, et al., 1996; Loucaides E M, et al., 2009; Perez D R, et al., 1995). As demonstrated in Figure 1C, both EGFPderived constructs and fusion proteins were expressed, with the expected sizes detected. Two RL-tagged constructs, following expression, exhibited good and comparable levels of luciferase activity, although fusion reduced the luciferase activity of RL-tagged constructs by approximately 10-fold in comparison with wild-type RL.
To examine influenza B polymerase PA and PB1 interactions, we first performed the LUMIER assay in a 12-well format. We cotransfected COS-1 cells with either EGFP-tagged constructs (B.PA and B.PB1) or an empty EGFP expression vector, and selected RL-tagged constructs or RL expression plasmids, respectively (Figure 1D). The association between the coexpressed proteins was assessed by performing a luciferase activity assay on anti-EGFP immunoprecipitates collected by centrifugation. Relative luciferase units (RLU) were calculated by subtracting background values obtained from cells transfected with an empty DNA vector, and were used to determine the presence of PPIs. Very low background luciferase activity (e.g., < 50 RLU) was detected in various anti-EGFP control immunoprecipitates (EGFP/ RL, EGFP/RL–B.PA, EGFP/RL–B.PB1, EGFP–B.PA/ RL, and EGFP–B.PB1/RL), demonstrating specificity of the LUMIER assay. A strong interaction between PA and PB1 (approximately 1005–1508 RLU) was observed regardless of the configuration of fusion proteins used (compare with EGFP–B.PA/RL–B.PB1 and EGFP– B.PB1/RL–B.PA in Figure 1D).
We also performed a traditional Co-IP assay to validate the influenza B PA–PB1 interaction (Figures 1E, 1F). We chose HA (YPYDVPDYA) epitope (-tagged PA over RL-tagged PA (a construct used in the LUMIER assay) because anti-HA antibody, not anti-RL, allowed us to precipitate the tagged PA fusion protein (data not shown). The EGFP–B.PB1 construct used to develop the LUMIER assay was also used in the Co-IP experiment. An interaction between PA and PB1 was demonstrated in cells expressing B.PAHA and EGFP–B.PB1 but not those expressing B.PAHA and EGFP, which is in good agreement with previous studies (Deng Q, et al., 2011a; Deng Q, et al., 2011b; Wunderlich K, et al., 2009; Wunderlich K, et al., 2010). This result further validates the utility of the LUMIER assay in detecting PA–PB1 interactions.
Next, we extended the PA–PB1 LUMIER assay to a 96-well microplate format amenable to a high-throughput assay (Figure 1G). In this experiment, following the lysis of cells expressing PA and PB1 proteins, protein A/ G magnetic beads were added to the wells containing the precipitate complexes, followed by automated magnetic bead washing. Washed cell lysates including magnetic beads were then assayed in a white 96-well plate for the presence of luciferase activity, indicative of a PPI. As shown in Figure 1G, background luciferase activity (i.e., < 20 RLU) was detected in control wells coexpressing EGFP–B.PA with empty RL vector. However, a robust interaction between PA and PB1 (approximately 400 RLU) was observed in wells expressing EGFP–B.PA with RL–B.PB1.
In summary, we have developed a simple, specific, and robust microplate-format PPI LUMIER assay. This methodology can be used to study PPIs that are critical to viral replication and pathogenesis. With further improvements such as automation, miniaturization, and quantitation, the LUMIER assay has the potential to become a powerful tool with which to study viral disease at a network level. The information derived using this assay will help with the design of novel inhibitors to combat viral diseases and in overcoming the resistances that are associated with current antiviral drugs.
-
This work was supported in part by the South Dakota Agricultural Experiment Station and the South Dakota 2010 Research Center, BCAAP (Biological Control and Analysis of Applied Photonics) Fund (3AH203 and 3SJ163 to FL).
All of the authors declare that they have no competing interest. This article does not contain any studies with human or animal subjects.

 DownLoad:
DownLoad: