










HTML
-
In recent years, owing to the fact that antibiotic-resistant bacteria have become more and more prevalent, there has been a resurgence of interest in the use of bacteriophages. However, bacteriophage therapy remains an underutilized option in modern medicine due to technical hurdles such as limited host range, narrow spectrum of bacteria affected by specific phages, side effects of bacterial lysis products, delivery, and so on (Sulakvelidze et al., 2009; Lu and Koeris, 2011). Advances in biotechnology and synthetic biology have showed many prospective of bacteriophage enzyme in antibacterial research (Walter, 2003; O'Flaherty et al., 2004). The use of purified bacteriophage enzymes has been considered as one key strategy to address the limitations of bacteriophage therapy (Uchiyama et al., 2001). A series of studies on purified bacteriophage enzymes and their activities toward Grampositive and Gram-negative bacteria have shown good therapeutic potential (Walter et al., 2003; O'Flynn et al., 2004).
Previous studies suggest that the gene gp17 from the K1F-like phage may play an important role in the processes of host range recognition and lysis (Kataoka et al., 2006; Chai et al., 2010). K1F-like phages are known to possess tail fiber protein with endosialidase activity that can degrade polysialic acid with high substrate specificity. These tail fiber enzymatic activities enable the phages to penetrate the bacterial capsular layer and are important determinants of the bacteriophage host range (Chai et al., 2010).
A novel phage, LSB-1, which can identify and specifically degrade the host bacterium enteroinvasive Escherichia coli 8401 (EIEC8401) (Chai et al., 2010), was discovered in sewage from a hospital in southwest China using the double agar plate method (Adams, 1959). EIEC is a bacterial pathogen that has caused many cases of dysenteric diarrhea in China. LSB-1 is a member of the Podoviridae family with a cystic form (50 ± 5 nm in diameter) with a short tail (60 ± 5 nm long) and double-stranded DNA of about 23 kb pairs (Chai et al., 2010). Bioinformatics analysis of gene gp17, the structure of enzyme activity site, and the genomic relationship between LSB-1 and other members of t h e phage T7 supergroup revealed that the gene gp17 of LSB-1 is very similar to K1F endosialidase (Broxmeyer et al., 2002) and encodes a tail fiber protein that can identify and lyze the cell walls of host bacteria (Chai et al., 2010). To provide another option for the therapeutic use of bacteriophage enzymes, here we set out to produce purified, recombinant endolysin gp17 from the LSB-1 phage and to examine the activity of the recombinant protein.
The genomic DNA of the LSB-1 phage obtained using a method described by Uchiyama et al.(2008a, 2008b) was approximately 23 kb (Figure 1A). The primers used in this study were designed based on the genome sequence and were synthesized by Sangon Biotech. The coding sequence of gp17 was amplified with primers, forward: 5'-TGAGCAGATCGGAGGCATGTCCACGAT TACACAATTCCCTTCAGG-3', and reverse: 5'-TGG TGGTGCTCGAGGTTATTCGCTGTAAACAAATCCG ATTCGC-3'. The length of the gp17 gene determined by PCR was about 2000 bp (Figure 1B). The PCR product was separated by electrophoresis in a 1% agarose gel and the gp17 product was gel purified. Subsequently, the purified gp17 PCR product was cloned into the Stu Ⅰ site of the expression vector pET300 to generate the pET300-gp17 recombinant plasmid, following the manufacturer's protocol for the In-Fusion™ PCR Cloning Kit. The recombinant pET300-gp17 plasmid was transformed into competent E. coli BL21 (DE3) cells for protein expression. The recombinant pET300-gp17 plasmid was confirmed by examining nucleotide sequences from positive trans formants on ampicillin agar plates; nucleotides were sequenced at Sangon Biotech. The gene encodes a protein of 713 amino acids with a theoretical molecular weight of about 78 kDa and an isoelectric point of 4.992. In our experiments, however, the recombinant protein was a pET300-gp17 fusion protein, so its molecular weight was about 104 kDa.
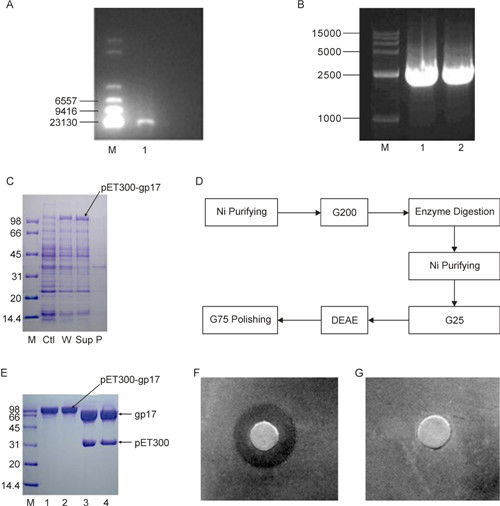
Figure 1. (A) Electrophoretogram of LSB-1 DNA. M: λ DNA Hind Ⅲ Marker; 1: DNA sample. This image was created using a Bio-Rad system and a 0.8% agarose gel after 1 h of electrophoresis. The band in lane 1 was the genome of phage LSB-1. (B) PCR of the target gene. M: 15, 000 bp Marker; 1 and 2: Target gene fragments. The target gene sample obtained by PCR was near the marker band at 2500 bp (gp17 has a size of 2139 bp). The PCR product was our target gene. (C) Expression of the recombinant protein pET300-gp17. M: Protein standard marker; Ctl: No IPTG; W: Whole bacterial extract; Sup: Supernatant of total E. coli soluble extracts after ultrasonication and centrifugation; P: Precipitate after ultrasonication and centrifugation. (D) Flow chart of the purification procedure of recombinant gp17 protein. (E) SDS-PAGE of pET300-gp17 cleaved by SUMO protease. M: Protein Standard Marker; 1: Sample under reducing conditions; 2: Sample under non-reducing conditions; 3: Digestion under reducing conditions of SDS-PAGE; 4: Digestion under non-reducing conditions of SDS-PAGE. (F, G) Bacteriostatic effect of recombinant lyase gp17 on the host of phage LSB-1, Escherichia coli 8401. (F) 5 μL lyase (2.38 mg/mL) incubated for 9 h; (G) 5 μL PBS buffer incubated for 9 h. The bacteriostatic circle occurred on treatment with the recombinant gp17 protein, but there was no bacteriostatic circle in negative controls.
E. coli BL21 (DE3) containing the recombinant expression plasmid was cultured in LB medium (10 mL; containing Amp and the final concentration is 100 μg/mL) at 37 ℃ overnight; half of the culture was then added to LB medium (100 mL) for another 3–4 h until the OD value (560 nm) was about 0.5. To overexpress the recombinant protein, the LB medium was supplemented with IPTG to a final concentration of 0.5 mmol/L, bacteria were cultured aerobically for another 10 h. To test for overexpression of the recombinant protein, the cultures were centrifuged (8, 000 × g; 5 min; 4 ℃) and the resulti ng cell pellets were dissolved in lysis buffer and ultrasonica ted. The supernatant, the precipitate from centrifugation (20, 000 × g; 4 ℃; 8 min), and the whole bacterial extract were electrophores ed to confirm overexpression of the protein by SDS-PAGE. SDS-PAGE in Figure 1C shows production of the recombinant protein (pET300-gp17) without IPTG induction was very low, while the production of the recombinant protein with IPTG induction was high in both bacterial total soluble extracts liquid and supernatant fluid after the lysis of recombinant bacteria. The pET300-gp17 level was lower in the precipitate than in the supernatant following ultrasonication of cells, demonstrating that the recombinant gp17 protein was soluble (Figure 1C). As judged by the protein molecular weight marker, the molecular weight of the recombinant protein (pET300-gp17) was about 100 kDa, close to the theoretical value of 104 kDa predicted by DNA Star software (DNA STAR, Inc. USA).
To get the purified recombinant protein, a series of procedure (Figure 1D) was performed to purify it and get rid of the label of pET300 (Figure 1E), and the purified recombinant protein was obtained and the concentration was determined to 2.38 mg/mL by the BCA method.
The activity of recombinant gp17 protein was determined by the Kirby-Bauer method (Bauer A W, et al., 1966). Briefly, 1.0 mL of EIEC8401 (2.0 × 108 CFU/mL) was spread on agar plates, on whi ch sterile paper with 5 μL recombinant protein (2.38 mg/mL) was placed after drying. PBS buffer (5 μL) was used as a negative control. Both samples were incubated at 37 ℃ to observe the effects. After incubating for 5 h at 37 ℃, a bacteriostatic circle formed on the agar plate that had been treated with recombinant gp17 (Figure 1F). These phenomena were not observed in the negative controls (i.e., PBS treated plates; Figure 1G). These results demonstrated that recombinant gp17 protein had a substantial bacteriostatic effect on the host of phage LSB-1, EIEC8401.
As the natural enemy of bacteria, bacteriophages have great potential in developing new strategies to solve the antibiotic resistance problem, although there are still many disadvantages to using bacteriophages directly (Sulakvelidze et al., 2009; Lu and Koeris, 2011). In this study, through a prokaryotic expression system, we successfully obtained purified, recombinant gp17 protein, a phage enzyme with high potential for resolving bacterial antibiotic resistance.
This study was based on a novel bacteriophage, LSB-1, which was isolated from sewage from a hospital (Chai et al., 2010) and is lytic against EIEC8401. The lyase coded by the gene gp17 is a tail fiber protein of the LSB-1 phage called acetyl neuraminidase, which is the key enzyme for identifying and dissolving the host bacteria. Protein three-dimensional structure analysis showed that the lyase of the LSB-1 phage consists of domains A and B. The N-terminus is located in domain A, the C-terminus is located in domain B, and β-sheet is the major structural component of the enzyme molecule. The enzyme active site is located in domain A and consists of Glu405, Arg415 and Arg487 (Chai et al., 2010). Having two active catalytic domains m ay decrease the possibility of resistance to this antimicrobial, because a pathogen would need two simultaneous mutations in the same cell wall to become resistant (Rodriguez et al., 2001). The recombinant gp17 lyase in this study had only one enzyme active site (in domain A), but domain B could be modified to have different catalytic activity. This could be another attractive aspect to exploit phage enzymes as potential antibiotics, because it has previously been shown that some individual catalytic domains can lyze cells in the absence of the complete protein (Becker et al., 2009; Sass and Bierbaum, 2007). Further analysis is required to confirm whether domain A only has the same activity.
In this study, we successfully expressed and purified the recombinant gp17 protein from the LSB-1 phage and also confirmed its bacteriostatic effect. Assays also showed that the recombinant enzyme was soluble and had significant lyase effects on the host bacterium, EIEC8401. A preliminary study demonstrated that the enzyme did not have inhibitory effects on other strains (unpublished data), which might indicate that the exclusive antibacterial effect of gp17 on EIEC8401 could have a special significance in practical application in bacterial therapy. To our knowledge, this is the first report of enteroinvasive E. coli phage lyase obtained by recombinase. Howev er, we have only tested preliminarily the antibacterial spectrum of the enzyme, and further studies should focus on factors affecting and improving the activity of the recombinant gp17 protein.
-
This research was supported by the Natural Science Foundation of China (30571637). All the authors declare that they have no competing interest. This article does not contain any studies involving human participants or animals performed by any of the authors.

 DownLoad:
DownLoad: