









HTML
-
Two novel betacoronaviruses, severe acute respiratory syndrome coronavirus (SARS-CoV) and Middle East respiratory syndrome coronavirus (MERS-CoV), have emerged in humans in this century, and both of which cause acute respiratory distress syndrome (ARDS) with high morbidity and mortality rates (Graham R L, et al., 2013). Both viruses are believed to originate in different bat species (Chan J F, et al., 2012; Ge X Y, et al., 2013; Lau S K, et al., 2005; Li W, et al., 2005; Zaki A M, et al., 2012), and have the potential to rapidly adapt and stably transmit to new species (Graham R L, et al., 2013). The patients with SARS are characterized by rapidly progressive pneumonia. The early symptoms are universal fever, myalgia, malaise and late symptoms are cough, dyspnea, tachypnea, pleurisy, and tendency of diarrhea. Most of the patients deteriorate to persistent fever, with increased dyspnea and oxygen desaturation (Peiris J S, et al., 2003). Severe histopathological alterations are evident in lung, including congestion, edema, diffuse alveolar damage, desquamation of pneumocytes, inflammatory infiltrates with hyaline membrane formation and interstitial thickening (DeAlbuquerque N, et al., 2006; Nicholls J M, et al., 2003). SARS patients show typical elevation of serum IFN-γ, IL-10, CXCL10/IP10, CCL5 and CXCL 8/IL-8 (Ward S E, et al., 2005).
Considering these extensive zoonotic reservoir and tendency to emerge as highly pathogenic human pathogens, it is therefore of high priority to establish valid animal models to understand virus-host interactions for drug and vaccine development. A line of animal models for SARS have been developed (Day C W, et al., 2009; DeAlbuquerque N, et al., 2006; DeAlbuquerque N, et al., 2006; Frieman M, et al., 2012; Leibowitz J L, et al., 2010; Ma X Z, et al., 2010; Roberts A, et al., 2007) ever since the first outbreak of SARS-CoV (Drosten C, et al., 2003; Ksiazek T G, et al., 2003; Osterhaus A D, et al., 2004; Peiris J S, et al., 2003), owing to the cross reactivity of spike protein with mouse ACE2 receptor orthologue. Closely recapitulating the clinical pathological manifestations in a progression is toward pneumonitis and diffused alveolar injury, but viremia is rare and transient. Moreover, SARS-CoV tends to adapt to mouse with elevated disease severity and mortality. MERS-CoV can transiently infect rhesus macaques with mild to moderate illness (Munster V J, et al., 2013), and common marmoset with severe pneumonia but absence of pulmorary fibrosis (Falzarano D, et al., 2014). Mice and hamsters are not permissive to MERSCoV due to lack of cognate receptor (Coleman C M, et al., 2014; de Wit E, et al., 2013). A humanized mouse model of MERS-CoV infection has been developed by transduction with an adenoviral vector expressing the human receptor of the virus, dipeptidyl peptidase 4 (Zhao J, et al., 2014). However, the requirements of ABSL3 level of containment and restricted license to conduct effective research further hinder the use of animal models for SARS-CoV and MERS-CoV infection.
Mouse hepatitis viruses A59 (MHV-A59) is a member of subgroup 2a of betacoronavirus, closely relating to human coronavirus HcoV-OC43 in the same subgroup and SARS-CoV in subgroup 2b (Graham R L, et al., 2013; Snijder E J, et al., 2003). A large body of evidence have associated pathologies with MHV-A59 infection, including hepatitis (Kim K D, et al., 2007; Weiss S R, et al., 2005; Zhao J, et al., 2008), autoimmune hepatitis-like disease (Aparicio J L, et al., 2011), thymus involution (Godfraind C, et al., 1995), hypergammaglobulinaemia (Coutelier J P, et al., 1987), and transient demyelination (Lavi E, et al., 1986). Whether MHV-A59 can infect respiratory tract and cause pathological manifestation similar to SARS-CoV infection remains elusive. Of note, MHV-A59 antigens can be detected in lung and disseminate to other tissues after neonatal Swiss mice are infected intranasally (Barthold S W, et al., 1984). Unfortunately, pathology has not been documented even in the immune compromised newborns.
To test whether MHV-A59 can infect the lungs, we intranasal inoculated C57BL/6 mice with MHV-A59. We found that MHV-A59 replicated in the lung and induced acute pneumonia, which closely mimicked ARDS by SARS-CoV and MERS-CoV in human lungs. This mouse model therefore may benefit further studies of pathogenesis of coronaviruses infection in human respiratory tracts.
-
Murine cell line 17Cl-1 was provided by Dr. K. Holmes (University of Colorado Health Sciences Center, US) and immortalized wild-type MEF cells in C57BL/6 background were original from ATCC, were maintained in the medium of Dulbecco's modified Eagle medium (DMEM) supplemented with 10% heat-inactivated fetal bovine serum (FBS) and antibiotics (100 μg/mL penicillin and 100 μg/mL streptomycin), at 37 ℃ in the incubator HERAcell® 240 with the presence of 5% CO2.
Mouse hepatitis virus A59 strain (MHV-A59) was propagated according to the method described previously (Schaad M C, et al., 1990). Briefly, the virus was propagated in 17Cl-1 cells followed by 3 cycles of freezing and thawing, the large debris was spun down and the supernatant was used as a stock solution. The titer of the virus was determined by plaque assay in 17Cl-1 cells exactly as described (Schaad M C, et al., 1990).
Sendai Virus (SeV) was obtained from Wuhan Institute of Virology, Chinese Academy of Sciences, China, and expanded in day 10 SPF embryonate eggs and titrated with hemagglutination assay (HA) as described previously (Zheng D, et al., 2008). The HA tier of SeV stock was 1 : 256, and for the infection of MEF cells 1 : 25 was used.
C57BL/6 mice were purchased from Beijing Laboratory Animal Research Center (Beijing, China). All mice were maintained in animal facilities under specific pathogen-free conditions. The handling of mice and experimental procedures were approved by the Animal Welfare and Research Ethics Committee of the Institute of Biophysics, Chinese Academy of Sciences.
-
4 week-old C57BL/6 mice were anesthetized intraperitoneally (i.p.) with pentobarbital sodium combined with chloral hydrate, and then inoculated intranasally (i.n.) with 10 μL of MHV-A59 virus at 1.5 × 105 or 1.5 × 104 plaque-forming unit (PFU), and control mice were inoculated i.n. with 10 μL of 17Cl-1 cell lysates in DMEM. The mice were monitored daily for health conditions. At intervals of 2, 5, 8, 11, and 30 days post-infection (dpi), 5 mice from each dose of infection group and 3 mice from control group were sacrificed. Lungs from infected and non-infected control mice were rinsed in ice-cold PBS, all tissues were separated into three portions for measurements of virus loads by plaque assays or RT-PCR, and for histological analysis (Hemmila E, et al., 2004).
-
To quantify the infectious virus particles in the lungs, portions of the lungs removed at necropsy were weighed and homogenized in DMEM with 10% FBS, and rapidly frozen and thawed for three times. Cell debris were removed by centrifugation, and the virus titers (PFU/g tissue) in the supernatants were determined by plaque assay on 17Cl-1 cells as described previously (Gombold J L, et al., 1993; Kim J C, et al., 1995; Lavi E, et al., 1984).
MHV-A59 N gene expression in the lung was analyzed by RT-PCR. Briefly, total RNA was extracted from 100 mg tissue with TRIzol reagent (Invitrogen, CA, USA), and 2 μg RNA, pretreated with 1 U of RQ1 RNase-free DNase (Fisher Scientific) to remove DNA contamination at 37 ℃ for 30min, were used for reverse transcription with oligo-dT primer(Promega). PCR reactions were then performed using the primers shown in Table 1 to detect the MHV-A59 N gene. PCR products (123 bp) were resolved by electrophoresis in 1.5% agarose gels and visualized a VersaDoc imaging system (Bio-Rad).
Description Forward 5' -3' Reverse 5' -3' MHV N gene CAGATCCTTGATGATGGCGTAGT AGAGTGTCCTATCCCGACTTTCTC L32 AAGCGAAACTGGCGGAAAC TAACCGATGTTGGGCATCAG 18s rRNA CCGCGGTTCTATTTTGTTGGT CTCTAGCGGCGCAATACGA IL-6 CACAGAGGATACCACATCCCAACA TCCACGATTTCCCAGAGAACA IP-10 CCAGTGAGAATGAGGGCCATA TCGTGGCAATGATCTCAACAC IFN-β AGCTCCAAGAAAGGACGAACAT GCCCTGTAGGTGAGGTTGATCT IFN-α4 ATGGCTAGGCTCTGTGCTTCC CATTGCAGAATGAGTCTAGGAG TNF-α GGTGCCTATGTCTCAGCCTCTT CGATCACCCCGAAGTTCAGTA IL-1β CAACCAACAAGTGATATTCTCCATG GATCCACACTCTCCAGCTGCA IFN-γ GCTCTGAGACAATGAACGCTAC TTCTTCCACATCTATGCCACTT IL-2 TGAGTGCCAATTCGATGATGAG AGGGCTTGTTGAGATGATGC IL-4 CATCCTGCTCTTCTTTCTCG CCTTCTCCTGTGACCTCGTT IL-5 GGGCCTTACTTCTCCGTGTA ACCTGAATAACATCCCAACCA IL-10 GGTTGCCAAGCCTTATCGGA ACCTGCTCCACTGCCTTGCT IL-12p40 GGAAGCACGGCAGCAGAATA AACTTGAGGGAGAAGTAGGAATGG Table 1. Primers used for current study
-
Lung tissues were removed from the mice at indicated time points, fixed in zinc formalin and processed as described previously (McCray P B, Jr., et al., 2007). For routine histology, sections were stained with hematoxylin and eosin, and the pathology of the lung was evaluated under microscope (Olympus CH30, Japan) by two independent technicians (Department of Pathology, China Agricultural University, Beijing).
-
Total RNA were extracted by Trizol from tissues or MEF cells as described above and quantitative real-time RT-PCR reactions were performed using SYBR Green Ⅰ (Molecular Probes, OR, USA) in a MyiQ system (BioRad, CA, USA) as described previously (Doyle S, et al., 2002). Relative levels of each gene were normalized to that of 18s rRNA. Primers for different genes were listed in Table 1.
-
All data were expressed as the mean ± standard deviations of the mean (SD). The results were analyzed statistically by using two-way ANOVA (Bonferroni post test) for body weight changes, and A Kruskal-Wallis test was used to analyze the differences in numbers of relative cytokines expression levels. Data with p ≤ 0.05 (*) and p ≤ 0.01 (**) were considered statistically significant.
Cells, Viruses and mice
Animal infection
Viral loads by plaque assay and RT-PCR
Histopathology evaluation
Cytokines profiles
Data analysis and statistical comparisons
-
To determine whether MHV-A59 can induce lung injuries in mice, we used sub-lethal doses of the virus (1.5 × 105 or 1.5 × 104 PFU/mouse)to intranasally (i.n.) infect 4 week-old C57BL/6 mice (n = 25 for each group). A higher dose (5 × 106 PFU and above) usually caused severe mortality thus not used in this study (data not shown). Mice infected by each dose began to show typical signs of illness at 3 days post-infection (dpi), including difficulty in walking, dormancy, ruffled fur and hunched posture. Infected groups lost appetite and reduced gain of body weights, as compared to the control mice (Figure 1A). By 6 dpi, most of mice (25 out of 30 mice) showed apparently respiratory distress, severe loss of appetite and emaciation. The body weights of infected mice start to drop rapidly at 5 dpi (1.5 × 105 PFU group) and 6dpifor (1.5 × 104 PFU group), respectively, which continued to decrease until to 9 to 10 dpi (Figure 1A). One mouse in 1.5 × 105 PFU group was deceased at 10 dpi.
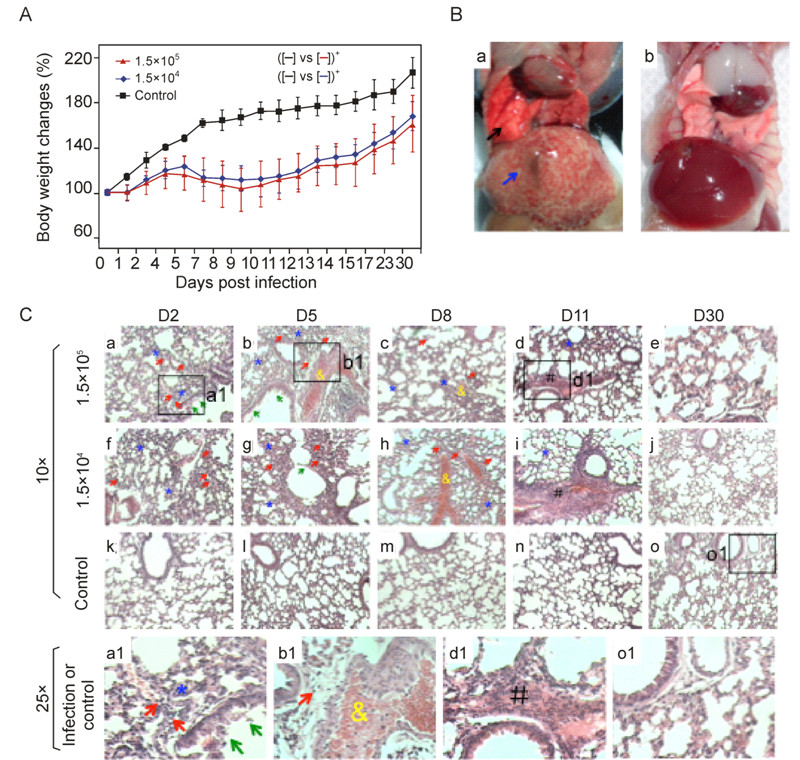
Figure 1. Pathologic alterations in C57BL/6 mice by intranasal infection of MHV-A59. A: Change of body weights were monitored daily after mice (n = 5 per group) were i.n. inoculated with different dose of MHV-A59, or 17Cl-1 cell lysates (n = 3) of sham infected (see methods) as the control. Two-way ANOVA post-hoc test. *, p < 0.05. B: Representative of sever congestive, hemarrhagic alterations in the lung (black arrow), and obvious necrotic foci in the liver (blue arrow) of 1.5 × 105 PFU group of mice (panel a). Control infection was shown in panel b. C: H & E staining of the lungs of infected (n = 5) and control (n = 3) mice at indicated time post infection. Indicated was inflammatory leukocyte infiltration in the alveolar walls (red arrows), alveolar walls thickening (blue stars), mucous epithelium fell off in the bronchioles (green arrows), hemorrhages in the small blood vessels (yellow ampersands) and fibrosis in the bronchial walls and the connection tissue between alveolar (black pound). Top panels a-e were 1.5 × 105 PFU group, panels f-j were 1.5 × 104 PFU group of infection, and panels of k-o were control mice. Higher magnification (25x) images from the infection (a1, b1 and d1) or control group mouse (o1) were shown in the panels at the bottom.
More importantly, analysis of the gross pathological alteration in the lungs showed obvious pneumonia at 5 to 8 dpi, and large areas of hemorrhages were evident in the lungs infected with 1.5 × 105 PFU MHV-A59 (Figure 1A). Both inoculation groups showed the similar progression from acute to chronic pneumonitis, with resolution at 30 dpi. On 2–5 dpi, both groups exhibited acute inflammatory response, including alveolar walls thickening (Figure 1C, panels a, b, f, g and a1, blue stars)and desquamation of mucous epithelia from the bronchioles (Figure 1C, panels a, b, g and a1, green arrows). Tissue damage exacerbated and peaked at 8–9 dpi, showing hemorrhages in the small blood vessels (Figure 1C, panels b, c, h and b1, yellow ampersands). The worst lung damage was correlated with the greatest loss of body weight (Figure 1A). As viral infection persisted, fibrosis in the bronchial walls and the connective tissues between alveolar septa became obvious (Figure 1C, panels d, i, and d1, black pounds). At the end of experiment (30 dpi), all the mice were able to recover from pneumonic injuries, and no significantly lesions were observed. The control group of mice did not show any lesions in the lungs (Figure 1C, panels k, l, m, n, o and o1).
Plaque assays of replicative virus in the lung homogenates showed that MHV-A59 at different inocula started to replicate in lung as early as 2 dpi (Figure 2A, panel a), with the virus titers reaching 5 × 105 and 2 × 106 PFU per gram of tissues, respectively. Higher titer of inoculum resulted in a faster viral clearance, with 20% and 60% of mice resolved on 5 dpi with the high and low dose of inoculum, respectively (Figure 2A, panel b). By 8 dpi, a very minute amount of virus could be detected in the lung (~100 PFU/g tissue in 20% of mice), and below the detection limit by 11 dpi for 1.5 × 104 PFU group. In parallel, RT-PCR analysis of MHV-A59 N gene in the lung showed the similar kinetics of virus replication and clearance (Figure 2B).
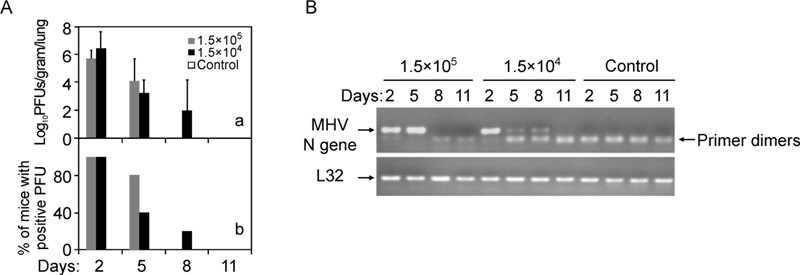
Figure 2. MHV-A59 replicated in the lung and produced infectious viral particles. A: Plaque assay measurement of production of infectious virions in the lungs (Log10 PFU/gram lung) after mice were i.n. infected with MHV-A59 (n = 5) or sham lysates (n = 3) for indicated time. The number of mice positive for detectable PFU was plotted over total number of inoculated mice (% of positive PFU). B: Detection of MHV-A59 replication at the indicated time post inoculation. MHV-A59 N gene in the lung homogenates (n = 5 for each time point post infection) from (A) was detected by standard RT-PCR amplification. L32 gene was used as a loading control.
Notably, as a hepatic virus, intranasal infection of MHV-A59 also caused fulminant hepatitis in C57BL/6 mice (Figure 1B), which is consistent with our previously report and others (Kim K D, et al., 2007; Lavi E, et al., 1986). Besides injuries in the lung and liver, the mice with intranasal infection of MHV-A59 also showed brain tissue damage under microscopy (data not shown), and it is consistent with the previous report (Lavi E, et al., 1986). Retention of gas in the intestines was also found in some of infected mice during necropsies (7 out of 20 mice). No obvious lesions were observed in the heart or kidney.
-
Histopathological changes would associate with inflammatory leukocyte infiltration in the lung. Both MHV-A59 inoculum groups exhibited significant lymphocyte infiltration in alveolar walls (Figure 1C, panels a, f, a1 and b1, red arrows), which included neutrophils, macrophages and eosinophils, among others. Furthermore, quantitative RT-PCR analysis showed that type Ⅰ inteferon response to MHV-A59 infection in the lung was rather limited, with transient induction of IFN-β only at 2 dpi, and null response of IFN-α4 within the course of study (Figure 3C, 3D). These results suggested that acute coronavirus infection in the lung was associated with a suppressed anti-viral type Ⅰ interferon response, in agreement with our previous results in vitro (Wang G, et al., 2011; Zheng D, et al., 2008). These results further support the notion that coronaviruses may have evolved an effective strategy to evade anti-viral innate immune response (Chen J, et al., 2007; Reghunathan R, et al., 2005; Wang G, et al., 2011; Zheng D, et al., 2008).

Figure 3. Proinflammatory cytokine response to MHV-A59 in the lung. Total RNA in the lung homogenates was extracted at indicated time post infection with 1.5 × 105 PFU MHV-A59 (n = 5) or sham infection (n = 3) as control. Real-time RT-PCR was preformed to measure indicated cytokine genes, with18s rRNA gene as internal reference. Kruskal-Wallis test. *P < 0.05; **, P < 0.01.
On the other hand, intranasal infection of MHV-A59 induced drastic up-regulation of IP-10 (Figure 3B) and IFN-γ (Figure 3G) in the lung, especially in the early phase of infection, suggesting involvement of macrophages and NK cells in the lung injury. This result agreed with our previous finding of NK cells and IFN-γsignal amplification in hepatopathologenesis(Kim K D, et al., 2007). The specific induction of IP-10 and suppression of IFN-βby MHV-A59 were further confirmed in vitro. Sendai virus (SeV) infection of murine embryonic fibroblasts (MEFs) could effectively trigger both IP-10 and IFN-βexpression (Figure 4C and 4D). In contrast, MHV-A59 could only stimulate IP-10 but not IFN-βexpression (Figure 4A and 4B). The hyper response of IP-10 and IFNγduring MHV-A59 infection was reminiscent of that in SARS-CoV infection (Huang K J, et al., 2005; Jiang Y, et al., 2005).
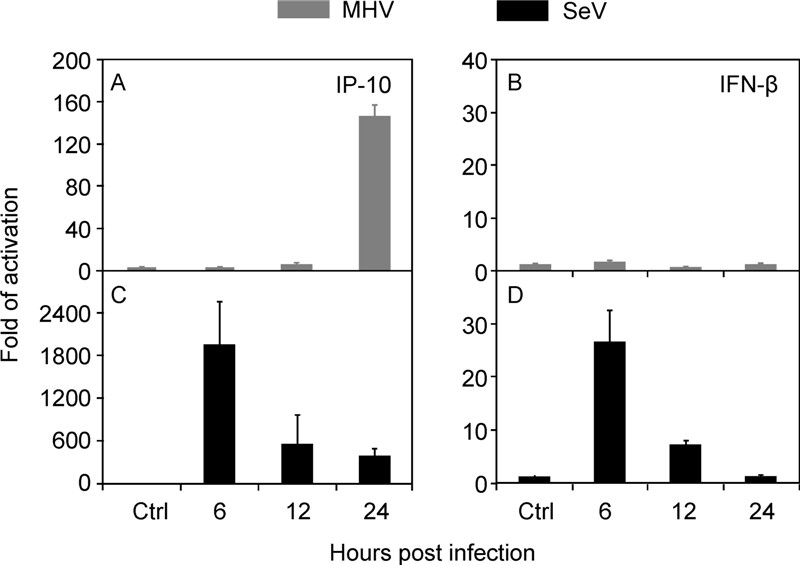
Figure 4. Differential activation of innate immune response. MEF cells (1 × 106) were infected with MHV-A59 (MOI = 5) or SeV (HA titer at 1:25) for indicated hours. The amount of IP-10 and IFN-βtranscripts was determined by real-time RT-PCR, after normalized to 18s rRNA. The data were average three independent experiments and expressed as fold of activation over that of mock infection(mean ± SD).
Other inflammatory cytokines involved in tissue damage and repair, such as TNF-αand IL-1β, were also significantly up-regulated in the lung (Figure 3E and F). TNF-α is a critical cytokine for MHV-A59 induced fulminant hepatitis, as we have previously reported (Kim K D, et al., 2007). IL-6 levels surged at 2 dpi and quickly came down to the basal level then after (Fig. 3A). Typical Th1 (IL-2, IL-5 and IL-12p40) or Th2 (IL-4 and IL-10) cytokines had no significant differences between the infected and control mice (Figure 3D, 3H-3L), suggesting that T cells activation would not play a major role in the lung injury. The lung of neonatal mice (lack of T cells) was also susceptible to acute infection by MHV-A59, with similar morbidity and mortality compared to adult mice (data not shown), support the notion that T cells may be dispensable to lung injury by coronavirus infection. Therefore, these results suggested that abnormal innate inflammatory activation and amplification in the lung would attribute to tissue injury and respiratory distress during acute coronaviruses infection.
Typical pneumonia was observed in MHV-A59 infection mice
Pro-inflammatory response to MHV-A59 infection
-
There still lacks suitable animal models to study pathogenesis of respiratory tract infection by SARS-CoV and MERS-CoV (Devitt E, 2013). This work is able to show that MHV-A59 by intranasal inoculation can lead to lung infection and severe pneumonic manifestations in C57BL/6 mice. The severity of MHV-A59 infection correlated with virus replication in the lung and causative tissue damage. Duration of lung infection reversibly correlates with the dose of inoculum, suggesting that a higher dose of MHV-A59 might stimulate more robust innate immunity to control the viral infection.
Recently studies revealed that mice intranasal infection of MHV-1 resulted in acute pneumonia in the lung (De Albuquerque N, et al., 2006; DeAlbuquerque N, et al., 2006; Leibowitz J L, et al., 2010; Ma X Z, et al., 2010), which were consistent with our observations by MHV-A59 infection. Although multiple strains of mice were applied for testing of MHV-1 infection, only A/J (De Albuquerque N, et al., 2006; Leibowitz J L, et al., 2010; Ma X Z, et al., 2010) followed by C3H/HeJ mice (De Albuquerque N, et al., 2006) were shown of higher susceptible to the lung disease of pneumonia. However, the immunological responses maybe altered in these mice with infection of coronavirus due to the immunological components deficient. Since multiple factors including IL-3, Naip5 (Naip5 (NLR family, apoptosis inhibitory protein 5), and complement factor C5 were deficient in A/J mice (JAX mice database), while C3H/HeJ mice was known for deficiency of TLR4 (JAX mice database).
Aberrant elevation of innate inflammatory response plays a critical role in lung injury and respiratory distress. In this sense, IP-10 may help chemoattracting NK cells (Biddison W E, et al., 1998; Farrar M A, et al., 1993; Loetscher M, et al., 1996)to lesions, as suggested in SARS (Huang K J, et al., 2005; Jiang Y, et al., 2005)and MERS patients (Chu H, et al., 2014). IFN-γ, primarily made by NK and T cells, is regarded as a pleiotropic cytokine immunomodulator that regulates a wide range of innate and adaptive immunity (Chesler D A, et al., 2002; Farrar M A, et al., 1993; Ruby J, et al., 1991). IFN-γis essential in development of coronaviral hepatitis (Kim K D, et al., 2007; Schijns V E, et al., 1998), MHV-JHM clearance (Marten N W, et al., 2001; Parra B, et al., 1999), and is one of major cytokine elevated in SARS patients (Huang K J, et al., 2005). MERS-CoV can also increase IFN-γproduction in infected cells (Lau S K, et al., 2013; Zhou J, et al., 2014), although little is known in MERS patients. Lastly, high mortality of ARDS patients (Meduri G U, et al., 1995; Meduri G U, et al., 1995), as well as SARS patents (He L, et al., 2006), also associates with up-regulated of IL-1β, TNF-αand IL-6 in the lung. Whether and how these cytokine contribute to pulmonary epithelial damage, the leading cause of acute lung injury and ARDS, remains to be determined. Therefore, acute lung injuries induced by i.n. infection of MHV-A59 in C57BL/6 strain will benefit further investigation into the pathogenesis of human coronavirus infections in respiratory tracts.
-
We thank Dr. Dahai Zheng for his contribution in the early phase of project development. Dr. K. Holmes (University of Colorado Health Sciences Center, US) to provide 17Cl-1 cell line and help optimize the protocol of MHV-A59 stock preparation. We also thank Dr. Ralph Baric (University of North Carolina, US) for the gift of MHV-A59 and guidance of virus infection. This work was supported in part by grants from the Natural Science Foundation of China (30430640, 31030031), and the National Basic Research Program of MOST (2004BA519A61, 2003CB514116, 2006CB504300) to H.T., and Natural Science Foundation of China (31400765) to J. D.
-
All the authors declare that they have no conflicts of interest. This article does not contain any studies with human or animal subjects performed by the any of the authors.
-
Conceived and designed the experiments: LS, GC and HT. Performed the experiments: ZY, GC. Analyzed the data: ZY, JD, JZ, XY and HT. Wrote the paper: ZY, JD and HT.

 DownLoad:
DownLoad: