









HTML
-
Human cytomegalovirus (HCMV) infects a large part of the population and has serious consequences for immunocompromised persons with AIDS or organ transplants and for newborns after congenital infection. HCMV is the leading viral cause of congenital birth defects (Britt W J, 1996). More than 30% of primary HCMV infections of pregnant women result in placental transmission and clinical syndromes. Congenital infection is not uncommon (Revello M G, et al., 2002; Revello M G, et al., 2002; Stagno S, et al., 1986). Of symptomatic newborns, about 12% die and half of the survivors develop mental retardation, vision loss, sensorial deafness, or a combination of any or all of these (Ramsay M E, et al., 1991). Interference strategies commonly target the early events of the replication cycle by using approved nucleoside analogs such as ganciclovir, the nucleotide analog cidofovir, and foscarnet. However, these can lead to resistance (Chou S, et al., 1997). In vitro anti-sense oligonucleotides against the HCMV immediate-early protein 2 (IE2) have proven effective (Anderson K P, et al., 1996), as has been targeting the UL36 and UL37 sites (Anderson K P, et al., 1996; Smith J A, et al., 1995). These attempts show that targeting the HCMV-IE part of the propagation cycle may be effective; however, none of these treatments are permissible or feasible in the potentially infected fetus. The lack of suitable treatment modalities has especially serious consequences for the congenitally infected fetus and for patients with impaired immune systems. The challenge is to find treatment modalities that do not depend on inhibition of the DNA replication process.
Viral gene expression is a result of the interaction of a virus and infected cells. In general, the virus needs to use cellular machineries to achieve successful gene expression. Viral gene regulation might be targeted to block viral replication. At the early time of cytomegalovirus (CMV) infection, 2 layers of viral gene regulation are critical: 1) IE gene regulation through the interaction of cellular factors with the major IE promoter (MIEP) and the MIE enhancer, and 2) activation of early genes by IE proteins (Stinski M F, et al., 2008). MIE gene regulation occurs at 3 levels: cellular and viral proteins incorporating the interaction with the enhancer and the promoter, the involvement of viral elements in introns, and gene-splicing regulation. After the immediate-early stage, the resultant IE proteins soon turn on the early gene expression program. One of the most studied early genes is early gene 1 (E1), also called UL112-113 (of HCMV) or M112-113 (of murine CMV) (Perez K J, et al., 2013). Many early gene products are required for viral DNA replication. Therefore, the studies on early gene expression regulation are also important in the developing of strategies against CMV via targeting the events before viral DNA replication.
There are about 7 animal CMVs (human, mouse, rat, guinea pig, monkey, chimpanzee, and rhesus), and they present a strict species specificity. HCMV can productively infect only humans (Jurak I, et al., 2006; Lafemina R L, et al., 1988; Tang Q, et al., 2006), so there is no straightforward animal model for HCMV infection. Murine CMV (MCMV) has many similarities with HCMV (Mocarski E S, Jr., Shenk, T., Pass R. F., 2006). First, both have the same replication cycles in host cells and cause similar diseases in their respective hosts. Next, they share similar genomic structures, and most of their respective gene products have similar functions. Last, HCMV and MCMV also have similar characteristics with respect to the immune response in vivo (Reddehase M J, et al., 2002). For that reason, MCMV is the second most widely investigated strain of CMV. Despite their similarities, apparent differences between HCMV and MCMV have been shown in many genes. Therefore, the mechanisms of gene regulation for HCMV cannot simply be applied to those of MCMV, and vice versa.
This review, aiming to elucidate the complicated interaction between CMV and host, will summarize the results of recent studies of both MCMV and HCMV. Focus will be placed on the very early events after the viral infection of permissive cells. Some experimental data are also, for the first time, shown in this review to further support the recently proposed hypothesis that intrinsic host cell defenses effectively limit or completely abrogate the spread of CMV infection—unless impeded by viral countermeasures. Understanding CMV early and immediate-early gene regulation is important for the development of better strategies to interfere with viral growth, especially before viral DNA replication.
-
Evolutionary variations have caused CMV to adapt to living in host cells and are often accompanied with alterations of the DNA sequence in favor of viral survival. MIE gene structure has been identified using a DNA sequencing program. Once CMV genomic DNA enters the nucleus, viral gene expression starts immediately. CMV MIE gene expression is controlled by the major immediate-early promoter (MIEP) and regulated by the CMV MIE enhancer (Stinski M F, et al., 2008). The CMV MIE enhancer locates upstream of MIEP and consists of several special DNA elements that recruit cellular transcriptional factors to form DNA-protein complexes (Figure 1). The formation of the complex might be the first line of interaction between CMV and host cells, and it happens in the nuclei of infected cells. These DNA elements (aka cis-elements) interact with different cellular transcription factors that include NF-κB, CREB/ATF, AP-1, RARRXR (Angulo A, et al., 1995; Angulo A, et al., 1996; Angulo A, et al., 1998), serum response factor, Elk-1, Sp-1, CAAT/enhancer binding protein, and the interferon-gamma activating sequence (GAS) (Meier J L, et al., 1996; Netterwald J, et al., 2005; Yang S, et al., 2005). Many of the cis-acting sites in the CMV MIE enhancer are repetitive.
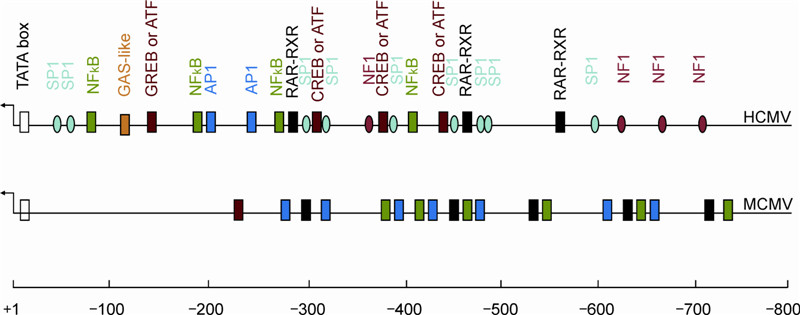
Figure 1. Comparison of the enhancers of HCMV with those of MCMV. The known consensus binding sites of cellular transcription factors in the major immediate-early enhancers are designated. Viral genes and promoter/transcription start sites are designated by arrows. The HCMV also has a unique region and a modulator, both of which are discussed in the text. The various transcription factor binding sites identified for HCMV and MCMV are designated.
It is still unknown which interaction is essential or important for MIE enhancer function. Most of the interactions between enhancer cis-elements result in activating effects on MIE gene expression; however, the HCMV MIE region contains 2 repressive elements, one that locates immediately upstream of the IE1/IE2 transcription start site and binds IE2 proteins (Cherrington J M, et al., 1991; Pizzorno M C, et al., 1990), and the one that binds a cellular protein (probably Fox-like repressors) immediately upstream of the TATA box of the divergent UL127 gene (Angulo A, et al., 2000; Lashmit P E, et al., 2004; Lundquist C A, et al., 1999). Additionally, evidence also shows that the silencing of the MIE enhancer is related to the variety of MIE gene expression in certain cell types, particularly when the cells are undifferentiated (Meier J L, 2001). The MIE enhancer has 2 functional components: the distal enhancer (−580 to −300) and the proximal enhancer (−300 to −39) relative to the transcription start site (+1). Deletion of either the distal enhancer or the proximal one negatively affects viral replication at low MOIs (Meier J L, et al., 2000). MCMV and HCMV have similar arrangements in the MIE gene area (enhancer, MIE gene, and divergent gene), except that transcriptional repression elements have not been found in the MCMV MIE gene.
Several aspects of the CMV MIE enhancer remain unclear: 1) Does the function of the CMV MIE enhancer play a pivotal role in regulating viral latency, reactivation, and pathogenesis? 2) Although many cellular transcription activators are identified as interacting with the specific site in the enhancer, their significance is unknown. Currently, which elements in the distal or proximal enhancer are required for virus replication in various cell types is also unknown. 3) The CMV MIE enhancer of one animal functionally differs from that of another. This dissimilarity needs to be demonstrated with experiments using animal models. 4) The requirement for particular HCMV MIE enhancer elements has not yet been assessed using viral mutants.
-
The host has developed various levels of defense against invading viruses, most prominently the adaptive and innate immune defenses, which protect the hosts from pathogenic infection (Tavalai N, et al., 2009). Does the host have any defensive response to the infected virus that is inside the cells? The short answer to that is perhaps. Nuclear repressors might function via interaction with the MIE enhancer. When a virus penetrates the host cell membrane, defenses based on the recognition of damaged or foreign proteins by the proteasome may reduce viral success. Once inside the nucleus, the foreign DNA may be recognized and destroyed by nucleases or may become chromatinized. Chromatinization can lead to silencing of the genome by a number of enzymatic complexes containing histone deacetylases (HDACs) (Guise A J, et al., 2013). Other nuclear-based regulatory mechanisms involved in silencing large stretches of the human genome may have evolved into mechanisms that suppress viral transcription and regulation. We have identified some of these intrinsic mechanisms.
Many ND10 (nuclear domain 10) components have been demonstrated to have a repressive effect on CMV gene expression and viral replication [reviewed by Saffert and Kalejta (Saffert R T, et al., 2008) and RiveraMolina et al. (Rivera-Molina Y A, et al., 2013)]. The first ND10 protein to be investigated for its role in HCMV gene expression and viral replication was Daxx. In the study that examined this role, Daxx was found to interact functionally with HCMV tegument protein pp71 (Hofmann H, et al., 2002; Ishov A M, et al., 2002; Saffert R T, et al., 2006; Tavalai N, et al., 2006). The Stamminger group (Tavalai N, et al., 2006) also investigated promyelocytic leukemia protein (PML) to see whether it could have any effects on viral gene expres sion or on viral replication. After comparing HCMV replication in PML-kd or hDaxx-kd cells with that in normal cells, Stamminger and his colleagues discovered that IE gene expression increased to a similar extent, regardless of whether PML or Daxx was depleted (Tavalai N, et al., 2009; Tavalai N, et al., 2006; Tavalai N, et al., 2011). Their experimental results suggest that PML and Daxx might function using different mechanisms to suppress HCMV replication; double-knockdown cells depleted of both PML and hDaxx support the additive enhancement of HCMV infection in the replication efficacy of HCMV compared to that of single-knockdown cells (Tavalai N, et al., 2011). Finally, they also found that the infection of SP100 knockdown (kd) cells with HCMV resulted in significantly increased plaque-forming ability (Adler M, et al., 2011).
Species-specificity is one of the major characteristics of CMV and is the primary reason for the lack of a mouse model for the direct infection of HCMV. It has been determined that CMV cross-species infections are blocked at the post-entry level by intrinsic cellular defense mechanisms (Cosme R C, et al., 2011; GarciaRamirez J J, et al., 2001; Jurak I, et al., 2006), but few details are known. We discovered that the ND10 of human cells is not disrupted by MCMV and that the ND10 of mouse cells is not disrupted by HCMV (Cosme R C, et al., 2011), although the ND10-disrupting protein, immediate-early protein 1 (IE1), also colocalizes with ND10 in cross-species infections (Cosme R C, et al., 2011). In addition, we found that the UL131-repaired HCMV strain AD169 (vDW215-BADrUL131) can infect mouse cells to produce IE and early (E) proteins but that neither DNA replication nor viral particles are detectable in mouse cells. Unrepaired AD169 can express only IE1 in mouse cells. In both HCMV-infected mouse cells and MCMV-infected human cells, the knocking-down of ND10 components (PML, Daxx, and SP100) resulted in significantly increased viral-protein production. Our observations provide evidence to support our hypothesis that ND10 and ND10 components might be important defensive factors against CMV cross-species infection.
More recently, we identified 2 other mechanisms that host cells use to defend viral replication. One of those mechanisms is found at the gene splicing level (Cosme R S, et al., 2009): We observed that polypyrimidine tract-binding proteins (PTBs) strongly repressed MIE gene production in cotransfection assays. In addition, we discovered that the repressive effects of PTB could be rescued by splicing factor U2AF. Taken together, the results suggest that PTBs inhibit MIE gene splicing by competing with U2AF65 for binding to the polypyrimidine tract in pre-mRNA. We conclude that PTB inhibits HCMV replication by interfering with MIE gene splicing through competition with U2AF for binding to the polypyrimidine tract in MIE gene introns. The second of the two mechanisms is associated with CTCF (Martinez F P, et al., 2014): We investigated the interaction of HCMV with the cellular chromatin-organizing factor CTCF. The results show that HCMV-infected cells produce higher levels of CTCF mRNA and protein at early stages of infection. They also show that CTCF depletion by short hairpin RNA results in an increase in MIE and E gene expression and an about 50-fold increase in HCMV particle production. We identified a DNA sequence (TTAACGGTGGAGGGCAGTGT) in the first intron (intron A) of the MIE gene that interacts directly with CTCF. The deletion of this CTCF-binding site led to an increase in MIE gene expression in both transient transfection and infection assays. The deletion of the CTCF-binding site in the HCMV bacterial artificial chromosome plasmid genome resulted in an about 10-fold increase in the rate of viral replication relative to either wild-type or revertant HCMV. Therefore, CTCF binds to DNA within the MIE gene at the position of the first intron to affect RNA polymerase Ⅱ function during the early stages of viral transcription.
-
To achieve replicative success, CMV has developed countermeasures against the cellular defenses. IE1, one of the most important proteins in the very early stage, helps the virus to evade host defenses, which are themselves accomplished via protein-protein interactions. IE1 is considered a promiscuous transactivator; however, all increases in protein production from viral or host promoters due to IE1 are modest, and the phrase "augmenting transcription" has recently been used specifically to describe the synergistic activation of early proteins by IE2. The modest increase in protein production due to IE1 is in stark contrast with the enormous effect of IE1 ablation on productive virus infection. Loss of IE1 has a disproportionate effect on viral replication, such that several orders of magnitudes more IE1-minus viral particles are necessary for plaque formation (Mocarski E S, et al., 1996). IE1 is not essential, but IE1 deletion mutants of HCMV require at least 1000 virus particles to overcome the defect, i.e., multiple hits are necessary for tegument proteins such as transactivator pp71 to activate the lytic cycle (Gawn J M, et al., 2002; Greaves R F, et al., 1998), and specific genes, such as that encoding dihydrofolate reductase, may require upregulation for sufficient nucleotide synthesis (Margolis M J, et al., 1995). We found that an IE1 deficiency can be rescued in permissive cells by the histone deacetylase inhibitor trichostatin A (TSA) (Tang Q, et al., 2003). Overall, the disproportionate effect of IE1 on viral replicative success appears to be due to the prevention or reversal of viral genome silencing rather than to minor additional viral protein production. Thus, interference with IE1 may block reactivation and attenuate any new infection.
IE1 disperses ND10. Like HSV-1, HCMV infection can also disrupt ND10, but the mechanisms of dispersing ND10 might be different. HSV-1 ICP0 induces the loss of the SUMO-1-modified forms of PML and the proteasome-mediated degradation of the PML protein (ChelbiAlix M K, et al., 1999; Everett R D, et al., 1998; Gu H, et al., 2003). However, in CMV-infected cells, PML is not degraded (Lee H R, et al., 2004; Tang Q, et al., 2003). For cytomegaloviruses (including MCMV and HCMV), IE1 has been identified to disperse ND10 by an as yet unknown mechanism, but it is not able to degrade PML (Ahn J H, et al., 1997; Korioth F, et al., 1996). HCMV IE1's induction of PML deSUMOylation, reported by Lee et al (Lee H R, et al., 2004), needs to be investigated for MCMV IE1. Obviously, interaction between ND10 with IE1 is not sufficient for IE1 to disperse ND10 because ND10 remains intact in cross-species infected cells (Cosme R C, et al., 2011). In our recent study of CMV cross-species infection, we infected MCMV into human fibroblast cells and infected HCMV into mouse fibroblast cells. We found MCMV IE1 colocalized with human ND10, and HCMV IE1 colocalized with mouse ND10, but ND10 is not dispersed (Cosme R C, et al., 2011).
IE1 interacts with ND10 proteins. A co-immunoprecipitation analysis of MCMV-infected 3T3 cells to identify repressor proteins that could be inactivated by IE1 revealed 3 such interacting proteins: HDAC2, Daxx, and PML, the latter confirming the interaction of MCMV IE1 with ND10. Several control proteins such a HSP70 and HSP25 were not immunoprecipitated. Using mouse cells in which either Daxx or PML was absent, we found that the interactions are independent from each other. We also found that IE1 can bring HDAC2 to ND10 at a very early stage in the infection, suggesting that the IE1 binding site is different from the Daxx binding site (Tang Q, et al., 2003). Whether the interactions are direct or within the context of a larger complex remains to be established.
Although the very low primary sequence homology of HCMV IE1 and MCMV IE1 suggests non-homology of the proteins, several functions are equivalent (e.g., binding PML and Daxx, dispersion of ND10, HDAC binding and the resultant augmentation of virus production), and functional domains in the respective gene structure are similar. It is not known whether these functional similarities of IE1 in the 2 species rest in the 3D structures or in similar interfaces. Comparison of the secondary structures shows a surprisingly similar predicted helicity in the first 400 amino acids of each protein and a similar spacing of shorter and longer helical sections (blocked areas in Figure 2). Moreover, a large proportion of the IE1 molecule forms a random coil, particularly the MCMV IE1 C-terminal region. It is necessary to perform a crystallographic or NMR structural analysis to reveal whether there is any real similarity between the 2 molecules that can account for their comparable functions.

Figure 2. Secondary structure of MCMV and HCMV IE1. Solid bars represent helical regions.
IE1 of HCMV and MCMV interact with HDAC complexes to inactivate HDAC. MCMV IE1 interacts with HDAC1 and 2 (Tang Q, et al., 2003), and HCMV IE1, with HDAC3 (Nevels M, et al., 2004). HDACs function only in several rather large complexes. HDAC3 is the essential silencing component in the silencing mediator for retinoic or thyroid (SMRT) repressor complex and nuclear receptor co-repressor (N-CoR) (Guenther M G, et al., 2001). The formed HDAC-associated complex functions as a gene silencer. In the IE1-bound complex, HDAC activity is inhibited (Tang Q, et al., 2003). If a cell silences an incoming virus through chromatization and deacetylation, then TSA should prevent such deacetylation and allow faster progression and higher production of progeny virus. Virus production increased ~8-fold in HCMV when the wild-type virus-infected host cells were at low multiplicities of infection, indicating that the competent virus is suppressed even in permissive cells. However, the strong activation of IE1-minus HCMV after exposure to TSA (Figure 3) suggests that a block in deacetylation activity rescues the IE1 deletion. These results, together with the observed suppression of HDAC1/2 by MCMV IE1 (Tang Q, et al., 2003), support the notion that IE1 sequesters a cell's silencing mechanism, and that this sequestration is analogous in both MCMV and HCMV.

Figure 3. Enhancement of CMV production after TSA exposure. PFU were determined on fibroblasts infected with wild-type or the IE1 deletion mutant at various times. Standard deviations are from 3 samples. Ordinate shows log 10 (adapted fromTang Q, et al., 2003).
Thus, the inhibition of IE1 activity appears to favor normal HDAC activity. Such activity would block transcription from the MIEP via silencing and essentially result in an IE1-minus phenotype, which cannot fully reactivate in the murine system (Kurz S K, et al., 1999; Reddehase M J, et al., 2002), i.e., it is important in organ transplant-dependent reactivation in humans. Our hypothesisis that IE1 binds and inactivates those HDACs responsible for gene silencing, thus preventing host cells from repressing viral transcription. It is important to de lineate IE1 binding sites for HDACs by deletion analysis and the expression of the smallest domain capable of binding. The minimal HDAC domain (or specific HDAC complex component) will be determined in the same fashion. Structural analysis of the interacting fragments will then reveal the shape of the interaction site, a prerequisite for the search for blocking agents. These analyses will identify the structure of the functional IE1 molecular domains that can be blocked by small molecules in order to inactivate IE1.
Inhibition of HDAC activity can synergistically enhance viral replication with IE2 in the IE2-producing cell line. To measure low levels of HCMV production, we generated a stable human cell line, MAMIE2 (Cosme R C, et al., 2011; Tang Q, et al., 2006a), in which IE2 expression is inducible with dexamethasone (Dex). As can be seen in Figure 4, treatment with either TSA or Dex produced an approximate doubling of plaques after infection with wild-type virus compared with that of uninduced cells, whereas the presence of both TSA and Dex increased plaque numbers more than 10-fold. This result suggests that the inhibition of HDAC (by TSA) and the upregulated expression of IE2 (by Dex) synergistically promote virus growth. The results also suggest that we have successfully produced a cell line with functional HCMV IE2 by suppressing normally detrimental IE2 expression during cell growth. Thus, we can produce inducible cell lines that more realistically recapitulate the temporal change in protein expression during infection. This may become especially important if cells adapt to the presence of a permanently expressed segregating protein such as IE1.

Figure 4. MAMIE2 cells infected with 0.001 PFU of HCMV and treated with 50 nmol/mL of the HDAC inhibitor TSA, or with dexamethasone, or with both compounds.
Evidence that MCMV can, with cooperation from HCMV IE1, breach the species barrier. Surprisingly, MCMV DNA replication proceeds in human cells. Despite evidence for MCMV DNA synthesis, no cytopathic effects were seen. Presumably, the block in particle production occurs at later stages of the replication cycle, and cells survive a rather large foreign invasion and viral DNA replication. Based on the initial assumption that human cells may produce virus but in quantities insufficient to sustain a second round of productive infection and plaque formation, we used time-lapse photography to monitor cultures infected with a green fluorescent protein (GFP) -producing MCMV (Henry S C, et al., 2000). Five cell lines were analyzed, including 2 that produced HCMV IE1. MCMV production was detected only in the IE1-producing human cell lines, and with kinetics as slow as that in HCMV-infected human fibroblasts. In the increasingly expanding MCMV plaque on the HCMV-producing astrocytoma (A1B) cell line, no apoptosis or cell lysis was evident (Tang Q, et al., 2006a). The same observations were made with the human IE1-producing fibroblast cell line ihfie1.3. MCMV plaque formation in 2 human cell lines expressing HCMV IE1 indicates that this effect is not cell type-specific, but rather is due to the presence of HCMV IE1 (Tang Q, et al., 2006a). It appears that there is no other principal block within a myriad of anticipated possibilities.
To identify the block in particle production in human cells, we produced an MCMV gene microarray covering 190 potential open reading frames (Tang Q, et al., 2006c). With this array, we detected new MCMV genes not previously identified as potential proteins and without equivalents in HCMV, as determined by sequence similarity. Preliminary findings indicate that MCMV replicating in human IE1-producing A1B cells produces signals for only 87 genes (compared to 160 genes in the murine 3T3 cells). Surprisingly, the number of genes in normal, non-producing human fibroblasts is only slightly lower than that in the producing A1B cells (Tang Q, et al., 2006c), which provides a reason for optimism regarding our efforts to identify and characterize the essential MCMV genes that require human HCMV IE1 for transcriptional activation.
IE1 exists in 2 complexes. IE1 is initially segregated to ND10 and binds to ND10-associated proteins, dispersing them within 3 h pi. Analyses of protein complexes by sedimentation, column chromatography, and co-immunoprecipitation require that complexes be soluble. Our sucrose sedimentation analysis (to assess the solubility state of the complexes) showed that the proteins of interest are soluble and present in sufficient quantities. They are at a mass indicative of their presence as complexes suitable for analysis. The bulk of IE1 resolved at size position ~200 kD, suggesting dimerization or heteromerization with another protein. PML and a breakdown product of Daxx, both of which are known to interact with IE1, appear in this region (Figure 5, lanes 7 to 9). In addition, the 36-kD isoforms of 112/113 have the same spread as the bulk of IE1, suggesting the need for an investigation of the binding to IE3 as well as to IE1. Since it is present in several complexes, HDAC2 is spread out as expected, with a distribution corresponding to a ~45-kD breakdown product of IE1. Stronger protease protection may need to be introduced during this type of analysis. At the larger sizes of IE1, i.e., ~670 kD, Daxx and the larger 112/113 isotype appear in a very tight range (lanes 2 to 4). Surprisingly, a rather large protein(lanes 2 to 4) reacts with antibodies to IE1 and 112/113. The large protein migrates to the same position as is occupied by normal Daxx and the larger 112/113 isoform. The presence of such a complex could be explained by covalent S-S binding due to air oxidation and incomplete reduction by 2-mercaptoethanol.
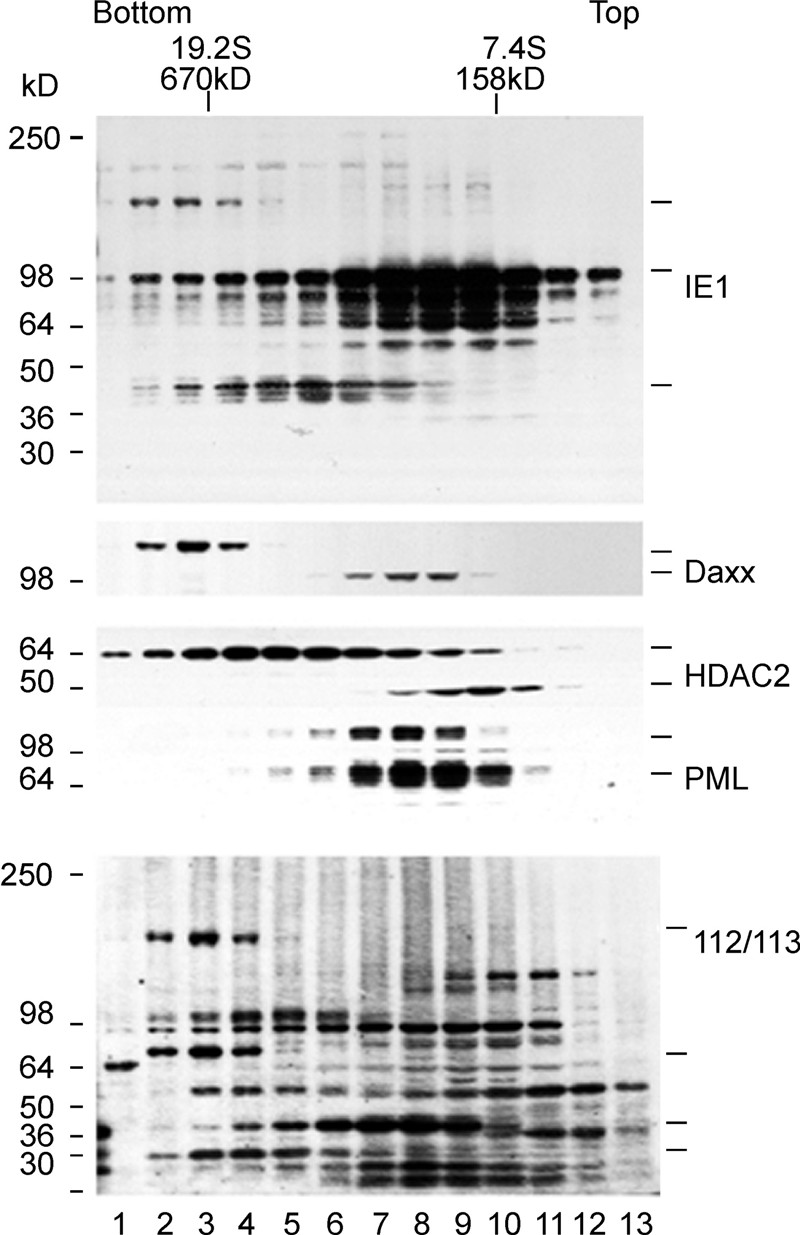
Figure 5. MCMV-infected 3T3 cells were harvested at 24 h pi and homogenized in 0.25 mol/mL sucrose in Tris buffer at pH 7.2, clarified at low rpm, and layered on a sucrose gradient before equilibrium centrifugation. Every 3rd sample was separated on SDS gels and, after blotting, sequentially tested with various antibodies, as indicated on the right. Rabbit anti-M112/113 antibodies reacted with additional cellular proteins. Bars on the right indicate specific bands.
-
Introduction. HCMV cannot successfully start the transcription sequence without the tegument-based transactivator pp71 (Baldick C J, Jr., et al., 1997; Bresnahan W A, et al., 2000). Thus, the function of pp71 represents a potential virus-specific site of interference. It has been shown that the HCMV pp71 is segregated to ND10 and the interaction of the C-terminal domain of Daxx with PML is the mechanism by which the cell segregates pp71 to ND10 (Ishov A M, et al., 2002). It has been also shown that pp71 directly interact with an N-terminal domain of the cellular repressor Daxx. We assume that the Daxx-dependent deposition/segregation of pp71 at ND10 reduces the availability of pp71 for viral transcription transactivation. In the case of an infection of relatively few viral particles, such segregation might have a defining effect on the activation of the infecting genome. The Daxx-dependent reduced availability of pp71 might attenuate viral production as effectively as a pp71 mutant would. Consistent with this hypothesis, Daxx-/-cells are very susceptible to low-level MCMV infection (60-fold more plaques than are found on mouse fibroblasts). Thus, the IE1 of HCMV or MCMV may have an equivalent function in undermining their respective host defenses through Daxx binding or destruction. The circumstantial evidence-based hypothesis of a defense and counterdefense remains to be proven. Our hypothesis is that the repressor Daxx inactivates the essential viral transactivator pp71 and that the inactivation of Daxx by IE1 rescues the essential pp71 functions. This presumption needs to be investigated with an eye towards fulfilling the following objectives: 1) Determine whether IE1 functions to counter the Daxx-based inactivation of pp71, and, if so, determine the mechanism of such a countermeasure. To assess whether the known IE1/Daxx interaction (Tang Q, et al., 2003) and the Daxx/pp71 interaction (Ishov A M, et al., 2002) are directly correlated in a functionally relevant way, it is important to establish whether these molecular interactions influence each other and, if so, how these interactions take place. 2) Determine whether the loss of the IE1-Daxx interaction but continued presence of the HDAC interaction impairs the virus. Countering the host's silencing of the viral genome and relieving the repression of the viral transactivator pp71 are 2 likely counterdefensive mechanisms of IE1.
Tegument-based pp71 transactivates viruses. It has been shown that the ND10-associated protein Daxx binds pp71 at its N-terminal region (Ishov A M, et al., 2002), whereas its C-terminal end interacts with the SUMO-modified PML (Ishov A M, et al., 1999). The HCMV tegument protein pp71, the product of the UL82 gene, activates the MIEP and other early promoters of this virus (Bresnahan W A, et al., 2000; Chau N H, et al., 1999; Liu B, et al., 1992). Moreover, pp71 can activate a number of heterologous promoters, both in the context of transient transfection and upon infection by a herpes simplex virus (HSV-1) mutant expressing pp71 (Homer E G, et al., 1999). The latter finding has led to the suggestion that pp71 may be the functional counterpart of HSV-1 tegument protein VP16, which transactivates the IE genes of this virus through interaction with a number of cellular proteins, including Oct-1. Transient transfection experiments also demonstrated that pp71 accumulates in the nucleoplasm (Hensel G M, et al., 1996) and dramatically enhances the infectivity of HCMV DNA (Baldick C J, Jr., et al., 1997), indicating that this tegument protein helps activate IE viral transcription before the production of HCMV IE transactivators in the context of infection. Growth of an HCMV mutant that does not express pp71 is severely restricted at a low multiplicity of infection, further pointing to the transactivation function of this tegument protein during the IE stage of infection (Bresnahan W A, et al., 2000). The adapter function of Daxx sequesters pp71 to ND10. We predict that the effect of this segregation makes pp71 unavailable for transactivation, and that IE1 segregation of the repressor Daxx can free pp71 to directly or indirectly activate IE and early promoters.
IE1-mediated offensive effects on nuclear defense
Pp71-mediated offensive effects on nuclear defense
-
CMV genomic DNA encounters cellular gene regulators in the nucleus; many of them are gene repressors such as HDAC and ND10 components PML and Daxx. As described above, pp71 and IE1 inactivate host cell gene repressors, including HDACs and ND10 proteins, and are required for viral gene activation and replication at low MOI (Ahn J H, et al., 1997; Cosme R C, et al., 2011; Hagemeier C, et al., 1992; Korioth F, et al., 1996; Mocarski E S, et al., 1996; Nevels M, et al., 2004; Tang Q, et al., 2003; Tang Q, et al., 2006b). The functional consequences of viral deposition at ND10 proved difficult to assess directly. Loss of pp71, which is necessary for HCMV deposition at ND10, resulted in reduced replication. However, this reduced replication was due to a lack of the necessary transactivation of various viral promoters. In the absence of Daxx, HCMV does not form its prereplication domain at ND10 (Ishov A M, et al., 2002), i.e., loss of Daxx abrogates viral deposition at ND10. The paradox of the virus's initiation of transcriptional and replicative functions at the site of repressive and interferon-upregulated proteins such as PML, Sp100, and Daxx has not been resolved.
-
This study was supported by a pilot grant from the Research Center for Minority Institutes (RCMI) program (2G12RR003050-24/8G12MD007579-27) (Q.T.), an American Cancer Society grant (RSG-090289-01-MPC)(Q.T), and NIH/NIAID SC1AI112785 (Q.T.). We are grateful to Bob Ritchie of the Ponce Health Sciences University/RCMI Publications Office (G12 RR003050/8G12MD007579-27) for his help with manuscript preparation.
-
All the authors declare that they have no competing interests. This article does not contain any studies with humanor animal subjects performed by any of the authors.
-
LT carried out the experiments, searched most of the references and participated in drafting the manuscript. QT designed the study and drafted the manuscript. Both authors read and approved the final manuscript.

 DownLoad:
DownLoad: