











HTML
-
Bacteriophages are powerful tools for investigating and manipulating their hosts (Fernandes et al., 2014). This holds particularly true for mycobacteriophages, which have facilitated the development of mycobacterial genetic systems and have generated tools for the clinical diagnosis of tuberculosis (Hatfull, 2010; Piuri et al., 2009). Good examples are the TM4-based genetic manipulation system (van Kessel et al., 2008), mycobacteriophage-derived peptides with killing activity against mycobacteria (Grover et al., 2014; Wei et al., 2013), and drug-resistant M. tuberculosis detection by fluoromyco-bacteriophages (Foongladda et al., 2014; Rondon et al., 2011). Recently, the emergence of multidrug-resistant (MDR-TB) and extensively drug-resistant (XDR-TB) strains of M. tuberculosis has become a major global health concern (Cully, 2014). The demand for new ways to overcome TB drug resistance has stimulated fresh research on mycobacteriophages and their lytic efficiency against their hosts (Oldfield and Hatfull, 2014; Sassi et al., 2014).
Currently, there are hundreds of genome sequences of mycobacteriophages available in GenBank. The availability of these sequences has helped in understanding the evolutionary relationships and genetic diversity among these phages. However, it appears that the mycobacteriophage population at large remains under-sampled, because new singleton phages with genomes entirely unrelated to known phages—as well as new relatives of previously-classified singleton genomes—can still be isolated. More work on the isolation and characterization of mycobacteriophages is therefore necessary (Hatfull et al., 2006; Hendrix, 2002). Mycobacteriophages with a high lytic efficiency are often chosen as genetic manipulation tools or TB-detecting tools, for example mycobacteriophage D29, which is used to detect M. tuberculosis in sputum and to determine its drug resistance.
In this study, we report the isolation and the complete genome of a novel mycobacteriophage, CASbig, which displays a plaque of around 1cm in diameter on a lawn of M. smegmatis mc2155 (Figure 1A). This virulent mycobacteriophage was isolated from a soil sample collected in Yanling County, Henan Province, PR China. As shown in Figure 1B, transmission electron microscopy (TEM) revealed that the CASbig phage has an icosahedral head (diameter 50 ± 2 nm) and a long, non-contractile tail (length 160 ± 5 nm) with transverse striations, ending in a small knob. The length of the tail includes the middle of the baseplate, and the head measurements were taken between opposite apices. These characteristics indicate that the phage belongs to the family Siphoviridae morphotypes.
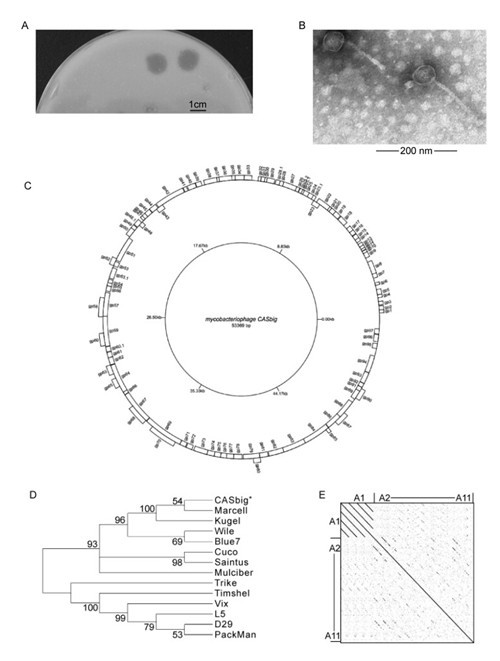
Figure 1. (A) Plaques formed by CASbig. (B) Transmission electron micrograph of phage CASbig. (C) Map of the mycobacteriophage CASbig genome. (D) Tree constructed by the maximum-likelihood method. The values at the nodes indicate the bootstrap scores using 1, 000 replicates. (E) Dot plot of mycobacteriophage cluster A genomes displayed using Gepard.
The genome of the phage was sequenced by means of a 454 sequencer (Genome Sequencer FLX System, Roche, US), which resulted in a single contig and showed that CASbig contains a double-stranded genome, 53, 369 nucleotides in length, with a GC content of 63.61%. Initial BlastN (http://blast.ncbi.nlm.nih.gov/last.cgi?PROGRAM=blastn&PAGE_TYPE=BlastSearch&LINK_LOC=blasthome) analysis of the CASbig genome showed that its closest relative at the nucleotide level, using the criteria stated in the Mycobacteriophage Database (http://phagesdb.org/), is mycobacteriophage Marcell, which belongs to the A1 subcluster. Using the Softberry program (http://linux1.softberry.com/berry.phtml?topic=virus&group=programs&subgroup=gfindv), the CASbig genome sequence was predicted to have 104 open reading frames (ORFs) with an average length of 534 nucleotides. Of the 104 ORFs, 17 are initiated with the s tart codon GTG, 86 with ATG, and 1 with TTG (gp24). Eighty-one of the ORFs have sequence similarity to other Mycobacteriophage Database entries, and 21 have been assigned functions, but one ORF (gp70) fails to match any genes. Although ORFs permit the prediction of potential genes, the analysis of further parameters, such as ribosome-binding sites, would help to identify the functions of these genes. The genome map of CASbig (Figure 1C) was obtained by means of GenomeVx software (http://wolfe.ucd.ie/GenomeVx/), which is a simple web-based tool for the creation of editable circular chromosome maps.
Bioinformatics analysis suggested that the genome of CASbig has a functional modular organization typical of tailed bacteriophages. No evidence of a lysogeny module was found in the genome. The 21 ORFs corresponding to potential genes with known functions could be classified into three functional groups: (1) nucleotide metabolism, DNA replication or recombination; (2) packaging and morphogenesis; and (3) lysis cassette.
Regarding the first group, 12 encoded proteins were identified as being involved in nucleotide metabolism, DNA replication or recombination. Those involved in nucleotide metabolism included an S-adenosylmethionine-dependent methyltransferase (gp8), exonuclease Ⅴ (gp22) and endonuclease Ⅶ (gp29). Hhpred analysis (http://toolkit.lmb.uni-muenchen.de/hhpred) showed that gp44, gp89 and gp97 had HNH endonuclease domains, indicating that these three proteins are all involved in nucleotide metabolism. Furthermore, one protein, gp27, was predicted to possess helicase activity, while gp42 was predicted to encode a DNA polymerase. A RecA-like NTPase, a primase and a DNA-protecting protein, encoded by gp27, gp33 and gp50, respectively, are indispensable for many aspects of DNA replication, and obviously play vital roles in the maintenance of DNA repair and genomic stability. Lastly, it appears that the integrase encoded by gp51 may be involved in recombination. This integrase (gp51) belongs to the serine recombinase family, which includes the transposon resolva ses and DNA invertases.
Phage packaging and morphogenesis is a complex process that requires temporal and coordinated activities of numerous proteins of both viral and host origin. In the CASbig genome, the genes involved are closely clustered and encoded by ORFs in the negative-sense strand. The terminase of the phage is encoded by gp84. Immediately downstream, gp83 encodes the portal protein. Two adjacent genes, gp93 and gp94, encode capsid proteins. The gp79 gene is predicted to encode the head structural protein. The gp59 gene, encoding a structural protein, is followed by a series of genes (gp64–83) involved in tail assembly, including the minor tail subunit (gp64 and gp67), tape-measure protein (gp69) and major tail subunit (gp83).
Following replication inside its bacterial host, the phage needs to lyse the host cell to liberate the progeny virions. Lysis of the host cell is a programmed event, and mycobacterium phages usually use two components for lysis – lysin A and lysin B. As in the mycobacterium phages Bxb1 and Marcell, in the CASbig genome, gp86 and gp88 encode lysin A and lysin B, respectively. Lysin B does not have the traditional lysin/holin structure of lysis cassettes found in bacteriophages; however, given the unusual lipid-rich cell wall of the host bacterium, the presence of this enzyme within the lysis cassette is not unexpected (Grover et al., 2014).
To address the evolutionary status of CASbig among the mycobacteriophages, we performed phylogenetic analysis using the tape-measure protein (TMP), which is the characteristic phylogenetic marker, conserved in mycobacteriophage genomes, and which is also the longest gene in these genomes (Smith et al., 2013). Since the genome sequence of CASbig, obtained using BlastN, showed a high degree of nucleotide sequence similarity to mycobacteriophages in cluster A in the Mycobacteriophage Database, the tree was simplified to include the representative members of cluster A mycobacteriophages. Phylogenetic analysis of the amino acid sequences of TMP in CASbig and other cluster A phages, including Marcell, Kugel, Wheeler, D28, L5, Vix, Wile, Cuco, Blue7, Timshel, Saintus, PackMan, Trike and Mulciber, was conducted using MEGA5 (http://www.megasoftware.net/megamac.php). Neighbor-joining and maximum-likelihood trees were constructed using 1000 bootstrap replicates. Both neighbor-joining and maximum-likelihood trees clearly placed CASbig on a branch adjacent to Marcell, in the A1 subcluster in the Mycobacteriophage Database (Figure 1D).
Gepard (http://bioinformatics.oxfordjournals.org/content/23/8/1026.full) is a useful and convenient tool for generating dot plots on a genome scale, which is used to assign mycobacteriophage genomes into clusters and subclusters. TMP was again selected in order to identify the known mycobacteriophage clusters and subclusters using a Gepard dot plot comparison. The dot plots suggested that CASbig is a member of cluster A1 and most closely related to mycobacterium phage Marcell (Figure 1E).
Nucleotide sequence accession number: The sequence data for phage CASbig were deposited in the GenBank database under accession number KC701493.
-
This work was supported by a grant from the State Key Laboratory of Virology (PR China) and Chinese Academy of Sciences (No. KJZD-EW-L02). The authors thank Dr Feifei Yin and Professor Zhihong Hu from the Wuhan Institute of Virology, Chinese Academy of Sciences for their help in sequencing the genome. All authors declare they have no competing interests. This article does not contain any studies involving human participants or animals performed by any of the authors.

 DownLoad:
DownLoad: