










HTML
-
As pathogenic agents of respiratory and enteral diseases, coronaviruses often cause serious disease in livestock and humans. For instance, the severe acute respiratory syndrome coronavirus(SARS-CoV), a member of the Coronaviridae family, led to more than 8400 cases of infection in 2003, with a 10% fatality rate(Ziebuhr, 2004). The Middle East respiratory syndrome coronavirus(MERS-CoV) has recently been identified highly virulent, with a mortality rate of over 35%(Zaki et al., 2012; Memish et al., 2013). SARS-CoV, classified under the genus β-Coronavirus, is a 5′-capped, positive-sense, single stranded RNA virus. Its genome is approximately 29.7 kb long and encodes 14 functional open reading frames(ORFs). The 5′ two-thirds of the genome contains two large overlapping ORFs, which encode two large polyproteins and produce 16 nonstructural proteins that play important roles during replication. The remaining 3′ one-third of the genome comprises 12 ORFs that encode four structural proteins as well as eight accessory proteins(3a, 3b, 6, 7a, 7b, 8a, 8b, and 9b)(Yount et al., 2005). These accessory proteins share no obvious homology to any known viral proteins. Therefore, their functions are still poorly understood. Previous studies have suggested that accessory proteins are important for virus-host interactions and are involved in protein incorporation into virus particles, apoptosis induction in the host cells, and modulation of the host's antiviral response(Lu et al., 2006; Chen et al., 2007; Kopecky-Bromberg et al., 2007; Schaecher et al., 2007).
Among all characterized accessory proteins, protein 6(P6, ORF6) has attracted the most attention owing to its structure and function. P6 has been shown to be highly conserved in SARS-related coronaviruses iso-lated from many different animal species, ranging from bats to humans(Guan et al., 2003). It is composed of 63 amino acids(aa), with a hydrophobic N-terminal region of ~40 aa and a hydrophilic C-terminal tail, and is localized in the cytoplasm and endoplasmic reticulum(ER)/Golgi membrane in infected cells(Zhou et al., 2010). P6 has been suggested to be required for optimal replication of SARS-CoV and to improve the productivity of mouse hepatitis virus(MHV)(Netland et al., 2007). It has also been reported to interact with nonstructural protein 8(nsp8) within the SARS-CoV replicase complex(Kumar et al., 2007) and to increase infection titer during early infection(Zhao et al., 2009). Functional studies in host cells have suggested that P6 can increase the rate of cellular gene synthesis(Geng et al., 2005) and induce Caspase-3-mediated ER stress, as well as Jun N-terminal kinase(JNK)-dependent apoptosis(Ye et al., 2008). Interestingly, P6 has been shown to be involved in modulating host antiviral responses. It inhibits both the synthesis and signaling of interferon-β(IFN-β) by suppressing the function of IFN regulatory factor 3(IRF-3), IFN-stimulated response element(ISRE) promoter, and the translocation of signal transducer and activator of transcription 1(STAT1)(Spiegel et al., 2005; Kopecky-Bromberg et al., 2007). Furthermore, it antagonizes STAT1 function by sequestering nuclear import factors on the rough ER/Golgi membrane(Frieman et al., 2007).
Here, we identified N-Myc(and STAT) interactor(Nmi) in a human cDNA library as a P6-interacting protein using the yeast two-hybrid system. Nmi was initially identified by a yeast two-hybrid approach with N-Myc as the bait(Bao and Zervos, 1996). Nmi is reported to be induced by interleukin-2(IL-2) and IFN-γ. Furthermore, by augmenting the association of CREB binding protein(CBP)/p300 coactivator proteins with STAT1/STAT5, it can enhance IL-2 and IFN-γ-dependent transcription(Zhu et al., 1999). Nmi has also been reported to interact with several other transcription factors, including all STATs, except for STAT2(Bao and Zervos, 1996; Zhu et al., 1999; Wang et al., 2013).
In the present study, we found that SARS-CoV P6 can interact with Nmi via the viral protein's C-terminal 54 to 63 aa region, leading to Nmi protein degradation through the ubiquitin(Ub) proteasome pathway(UPP). Based on our results, we propose that SARS-CoV P6 can mediate cellular Nmi degradation and thus favor the survival of SARS-CoV in host cells by eliminating Nmi-stimulated downstream IFN immune signaling.
-
Anti-FLAG(F4045) and anti-HA(H9658) antibodies were purchased from Sigma-Aldrich(St. Louis, MO, USA). Goat anti-mouse secondary antibody(sc-2005), anti-β-actin antibody(sc-130656), and Protein A/G PLUS Agarose(sc-2003) were purchased from Santa Cruz Biotechnology, Inc.(Santa Cruz, CA, USA). Cy3-labeled goat anti-rabbit IgG antibody(P0183) was purchased from Beyotime Institute of Biotechnology(Jiangsu, China). MG-132(M7449) was purchased from Sigma-Aldrich. Anti-Ub mouse monoclonal antibody, FK2(BML-PW0150-0025), for the detection of mono-and polyubiquitinylated conjugates, was purchased from Enzo Life Sciences, Inc.(Farmingdale, NY, USA).
-
pCAGGS-P6-HA and pEGFP-P6 plasmids were gifts from Dr. Stanley Perlman. The PCR primers employed in the synthesis of the constructs are shown in Table 1. For yeast two-hybrid screening, P6 and P6-N(40 aa) were PCR amplified and inserted into the EcoR I and Sal I restriction sites of the pGBKT7 vector. P6-C(24 aa) was PCR amplified and inserted into the Nde I and Sal I restriction sites of the pGBKT7 vector. For mammalian expression, pEF-FLAG-P6 was constructed by inserting PCR amplified P6 into the Xho I and EcoR I restriction sites of the pEF-FLAG vector. For P6 mutant(M1, M2, and M3) expression, the PCR products were digested with Xho I and EcoR I and cloned into the pEF-FLAG vector. The human Nmi sequence was amplified by PCR using cDNA from human embryonic kidney(HEK)293T cells as the template and was inserted into the SalI and NotI restriction sites of the pRK-HA vector or inserted into the BamH I and Sal I sites of the pRK-FLAG vector. Constructs were confirmed by sequencing.

Table 1. The primers used in the study
-
MatchmakerTM GAL4 Two-Hybrid System 3 was purchased from Clontech Laboratories, Inc.(Mountain View, CA, USA), which includes a human cDNA library, a yeast strain AH109, empty vectors pGBKT7, pGADT7 and control vectors pCL1, pGADT7-T and pGBKT7-53. Yeast two-hybrid screening and related assays were performed according to the manufacturer's instructions. For fluorescence imaging, post-transfection cells were incubated with polyclonal mouse anti-HA antibody for 2 h and Cy3-labeled goat anti-mouse IgG for 1 h and then observed using laser confocal microscopy(FV1000, Olympus, Tokyo, Japan).
-
Assays were performed as described previously(Chen et al., 2013) with the following minor modifications: for ubiquitination immunoprecipitation, cell extracts were denatured by boiling for 5 min with lysis buffer(1% Tritox X-100, 20 mmol/L Tris-HCl, pH 7.4, 150 mmol/LNaCl, 1 mmol/L EDTA, 1 mmol/L PMSF, 1 mmol/L DTT and protease cocktail) containing 1% sodium dodecyl sulfate(SDS). Next, the denatured extracts were diluted with lysis buffer at 10× the extract volume and were immunoprecipitated with mouse anti-HA antibody. Protein ubiquitination was analyzed by western blotting with the anti-Ub antibody, FK2.
-
Total RNA was extracted from cultured 293T cells with TRIzol Reagent(Invitrogen, Carlsbad, CA, USA) according to the manufacturer's instructions. Gene transcripts were semi-quantified by PCR and DNA electrophoresis, and normalized to the housekeeping gene GAPDH. Primers for RT-PCR of Nmi were: sense 5′-GCTCTAGAATGGCAACGGATGAAAAGGA-3′; anti-sense 5′-CAAGCTTCTATTCTTCAAAGTATACT ATGTGA-3′. The primers used for RT-PCR of GAPDH were: sense 5′-GACATCAAGAAGGTGGTGAA-3′; anti-sense 5′-TGTCATACCAGGAAATGAGC-3′.
Antibodies and chemical reagents
Plasmids
Yeast two-hybrid screening and fluorescence imaging
Immunoprecipitation and western blot analysis
Reverse Transcription(RT)-PCR
-
As a highly conserved accessory protein in SARSrelated coronaviruses, P6 has attracted much interest from researchers. Previous studies have shown that P6 is involved not only in virus replication and infection but also in host gene regulation and immune responses(Spiegel et al., 2005; Kopecky-Bromberg et al., 2007; Kumar et al., 2007; Netland et al., 2007; Zhao et al., 2009). To further explore the function of P6 and identify its host-cell binding partners involved in SARS-CoV infection, we employed a yeast two-hybrid system to screen a human spleen cDNA library purchased from Clontech Laboratories, Inc. for proteins that interact with P6. Fourteen positive clones were characterized and further analyzed to eliminate repeated sequences. Subsequently, eight host proteins were identified and these are shown in Table 2.

Table 2. Potential host cell proteins inter racting with severe acute respiratory syndrome coronavirus (SARS-CoV) protein 6 (P6)
From these potential P6-interacting proteins, Nmi was selected for further study. Nmi has been previously shown to be induced by IFN and to regulate IFNdependent transcription(Zhu et al., 1999). Previous studies have evidenced that P6 can inhibit the gene expression induced by ISRE promoter after Sendai virus(SeV) infection(Kopecky-Bromberg et al., 2007), although the mechanism underlying this process remains unclear. We speculated that P6 might regulate IFN-dependent transcription through interactions with Nmi to inhibit Nmiregulated IFN signaling.
In our yeast two-hybrid experiments, we identified two positive clones of Nmi that interact with SARS-CoV P6(prey D and I, Figure 1A). Both contain the Nmi 1-260 aa region, suggesting that the interaction domain of the protein might be located in this region. We also cloned the hydrophobic N-terminal region(P6-N) and hydrophilic C-terminal tail(P6-C) of P6 with yeast two-hybrid BD vectors and tested their interacting properties with Nmi. The results showed that the 24 hydrophilic C-terminal residues of P6, but not the 40 hydrophobic N-terminal residues, could interact with Nmi(Figure 1B). Therefore, we concluded that the C-terminal hydrophilic tail of SARS-CoV P6 facilitates the protein's interaction with host Nmi protein.
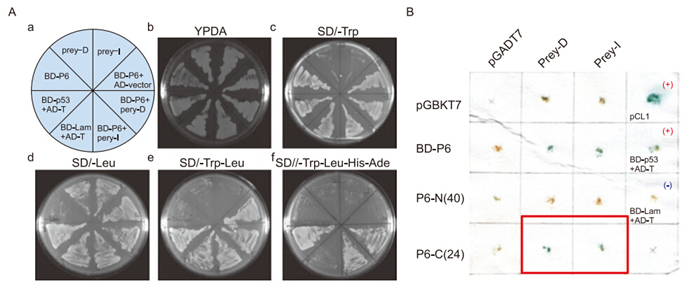
Figure 1. Severe acute respiratory syndrome coronavirus (SARS-CoV) protein 6 (P6) interacts with N-myc (and STAT) interactor (Nmi) in yeast and in vivo. (A) Yeast strain AH109 was transformed with plasmids and plated onto synthetic minimal medium (SD) plates as indicated in A. Prey-I: AD-Nmi 1–260 aa. Prey-D: AD-Nmi 1-267 aa. Positive control: BD-p53 + AD-T. Negative control: BD-lam + AD-T. (B) Positive colonies were retested using the X-Gal assay. The blue color shows positive interaction. P6-N (40 aa): BD-P6 1–40 aa. P6-C (24 aa): BD-P6 40–63 aa. Positive controls: pCL1 and BD-p53 + AD-T. Negative control: BD-lam + AD-T. AD: activation domain, BD: binding domain.
To determine whether P6 also interacts with Nmi in vivo, we cloned P6 and Nmi into eukaryotic vectors and transfected these plasmids into 293T cells. The interaction between Nmi and P6 was detected by colocalization analysis using immunofluorescence. Using confocal microscopy, we observed that the expression of P6 and Nmi, shown by green and red fluorescence, respectively, was colocalized. This was represented by the merging of the fluorescence colors into yellow fluorescence at particular spots within cells(Figure 2A, 2B), indicating that P6 could interact with Nmi in vivo. In addition, although single transfection of P6 exhibited a similar pattern to co-expression, since green spots were observed in both instances(Figure 2A, 2B), P6 seemed to modify the subcellular localization of Nmi. In single transfection, Nmi was spread evenly throughout the cytoplasm(Figure 2B); while when co-expressed with P6, Nmi aggregated in some areas to colocalize with P6(Figure 2A). These results suggest that through their interaction, P6 could recruit Nmi.

Figure 2. Colocalization of severe acute respiratory syndrome coronavirus (SARS-CoV) protein 6 (P6)-GFP and N-myc (and STAT) interactor-hemagglutinin Nmi-HA in human embryonic kidney 293T cells. (A) 293T cells were transfected with 0.5 µg pEGFP-P6 and 0.5 µg pRK-Nmi-HA. 24 h post-transfection, cells were incubated with polyclonal mouse anti-HA antibody for 2 h and Cy3-labeled goat anti-mouse IgG for 1 h and observed under a fluorescence microscope. (B) 293T cells were transfected, fixed, and labeled as in (A). Cells were observed under a fluorescence microscope. GFP: green fluorescent protein.
-
FLAG-tagged Nmi, either alone or together with HAtagged P6, was(co) transfected into 293T cells and their respective protein levels were determined using western blot analysis. The results showed that P6 could promote Nmi protein degradation in a dose-dependent manner(Figure 3A). To further confirm that P6 regulates Nmi at the protein level, RT-PCR was adopted to show that P6 does not affect Nmi mRNA production(Figure 3B). These results indicated that P6 could regulate Nmi protein levels by promoting Nmi degradation. To examine whether P6-promoted Nmi degradation is dependent on the UPP, proteasome inhibitor MG-132 was used to treat the cells and Nmi ubiquitination was examined by HANmi immunoprecipitation followed by anti-Ub western blot analysis. The results showed that the P6-mediated Nmi degradation could be partially recovered by MG-132 treatment(Figure 3C, 3D), and that Nmi ubiquitination was increased from control levels by P6 cotransfection and was further increased by MG-132 treatment(Figure 4A). These results indicated that P6 could stimulate Nmi ubiquitination and thus promote Nmi degradation through the proteolytic UPP.

Figure 3. Severe acute respiratory syndrome coronavirus (SARS-CoV) protein 6 (P6) mediates ubiquitin-dependent proteosomal degradation of N-myc (and STAT) interactor (Nmi). (A) Western blotting (WB), with indicated antibody, for human embryonic kidney 293T cells transfected with 2 µg pRK-Nmi-FLAG together with increasing amounts (0.2 µg, 0.5 µg, 1 µg, 2 µg) of pCAGGS-P6-HA. Cell extracts were analyzed 24 h post-transfection. (B) Reverse transcription-PCR results for 293T cells transfected with 2 µg pRK-Nmi-FLAG together with 2 µg pCAGGS-P6-HA, pEGFP-P6, or their respective empty vectors pCAGGS and pEGFP. Total RNA was extracted 48 h posttransfection. (C) Immunoblots, with the indicated antibodies, of 293T cells transfected with 2 µg pCAGGS-P6-HA and 2 µg pRK-Nmi-FLAG and treated with dimethyl sulfoxide (DMSO)/20 µmol/L MG-132 for 4 h before harvesting of cell extracts. (D) The relative expression of Nmi (Figure 3C, first panel, lane 1, 4, and 5) was calculated and graphed with grey intensity analysis. The bar graphs indicate mean values ± SD of triplicate samples from three independent experiments. * indicates P < 0.05.
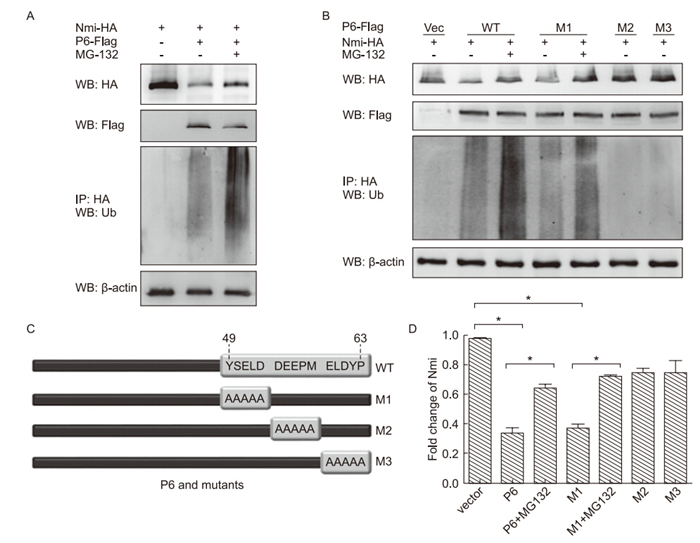
Figure 4. The activity N-myc (and STAT) interactor (Nmi) degradation-mediating activity of severe acute respiratory syndrome coronavirus (SARS-CoV) protein 6 (P6) mutants. Human embryonic kidney 293T cells were transfected as indicated, and treated with dimethyl sulfoxide (DMSO)/20 µmol/L MG-132 for 4 h. Cell extracts were harvested and analyzed by immunoprecipitation and western blotting with the antibodies indicated. (A) 6 µg pRK-Nmi-HA and 4 µg pEF-FLAG-P6 were transfected into 293T cells. (B) Diagram of P6 mutants. (C) 6 µg pRK-Nmi-HA was transfected with 4 µg pEF-FLAG-P6 wild type (WT) or M1, M2, or M3 mutant. (D) The relative expression of Nmi (cells in Fig. 4C, first panel) was calculated and graphed with grey intensity analysis. The bars indicate mean value ± SD of triplicate samples from three independent experiments. * indicates P < 0.05.
-
Based on our results of the yeast two-hybrid system and structural analysis, we predicted that the C-terminal of P6 might play the key role in its inhibitory activity on Nmi protein. Three P6 mutants with alanine mutations on amino acids 49 to 53, 54 to 58, and 59 to 63 were constructed and designated the names M1, M2, and M3(Figure 4B). Their capacity to induce Nmi degradation and ubiquitination was examined and compared with wild type(WT) P6. The results showed that M1 demonstrated similar activity to WT P6; enhanced Nmi ubiquitination and degradation. However, both M2 and M3 had no effect on Nmi ubiquitination and degradation(Figure 4C, 4D). These results further confirmed our conclusion that SARS-CoV P6 functions to promote Nmi ubiquitination and degradation through interacting with Nmi. In addition, we found that the C-terminal 54 to 63 aa of P6 are critical for its functions.
Identification of Nmi as a host protein that potentially interacts with SARS-CoV P6
SARS-CoV P6 regulates the degradation of Nmi through the UPP
P6 regulates Nmi ubiquitination and degradation via a small region residing on P6's C-terminus
-
To evade host innate immune responses, viruses have evolved various mechanisms to inhibit or modify host innate immune signaling pathways. IFNs, as cytokines with great importance during host innate antiviral immune responses against invading pathogens, have become the target of a diverse range of viruses(Garcia-Sastre and Biron, 2006). In many cell lines, SARS-CoV does not induce type Ⅰ IFN production and seems to encode several proteins to suppress IFN signaling(Garcia-Sastre and Biron, 2006). A previous study showed that P6 could bind to import factor karyopherin α2(KPNA2) and func-tion to disrupt nuclear import complex formation. Thus, P6 prevented the nuclear translocation of STAT1 and the downstream expression of STAT1-activated genes during IFN signaling, leading to failure of the host to establish an antiviral state(Garcia-Sastre and Biron, 2006). Here, we propose a new mechanism by which SARS-CoV restricts IFN antiviral signaling through P6 activity. Based on our findings, we suggest that P6 may interact with host Nmi protein and induce its degradation, thus blocking the host antiviral immune response by inhibiting Nmi-stimulated activation of ISRE. Both of these mechanisms may be employed by P6 to suppress IFN signaling, but each has different target factors and steps during the signaling pathway. One mechanism specifically targets Nmi to induce its destruction, suppressing Nmienhanced IFN signaling; the other targets general nuclear import processes to block STAT1 nuclear translocation and hence downstream IFN signaling. It is possible these mechanisms represent two complementary methods to suppress IFN pathway induction. Interestingly, we found that the C-terminal 54 to 63 aa of P6 were critical for P6-mediated Nmi ubiquitination and degradation, while the same region was also critical for P6 to block nuclear import, implying a link between these two functions of P6 during IFN signaling pathway suppression. Whether this connection is based on functional interplay or a co-regulating factor requires further investigation.
SARS-CoV antagonizes the IFN signaling pathway by blocking the nuclear import machinery, this mechanism is employed by diverse viruses. Ebola virus VP24 can also restrain the nuclear translocation of STAT1 by binding to import factor karyopherin α1(KPNA1) and blocking the nuclear import complex(Reid et al., 2006). The capsid protein of Venezuelan equine encephalitis virus(VEEV) is able to form a complex with the host proteins importin α/β1 and CRM1; thus, blocking nuclear trafficking and modulating host transcription(Lundberg et al., 2013). Similarly, there are many cases in which viruses utilize the ubiquitin proteasome system to degrade specific host proteins in order to suppress the host's innate immune system(Randow and Lehner, 2009). In the present study, we proposed that SARS-CoV P6 could promote host Nmi degradation, thus inhibiting the downstream IFN signaling pathway. However, it is not yet known whether Nmi can undergo ubiquitin-dependent proteosomal degradation during normal cellular processes. Further research is needed to reveal the role of ubiquitin modification in the functioning and regulation of Nmi in both the absence and presence of viral invasion.
In this study, the interaction between P6 and Nmi was elucidated using yeast two-hybrid screening and was confirmed by immunofluorescence. We showed that P6 may influence the subcellular localization of Nmi and is able to promote Nmi ubiquitination and degradation. However, we failed to detect any interaction of P6 with Nmi when conducting immunoprecipitation. One possible reason for this is that P6 localizes to the Golgi membrane, through its N-terminal 41 aa, but Nmi is mainly present in the cytoplasm(Zhou et al., 2010). When Nmi interacts with P6 at its C-terminal cytoplasmic region, the interaction might be weak and could therefore be disturbed during the membrane disruption induced by the lysis buffer. Another possibility is that the interaction between P6 and Nmi is transient and becomes rapidly disassociated after association, leading to negative results in the immunoprecipitation experiments. We also failed to obtain purified P6 for in vitro binding experiments. The question of whether the P6-Nmi interaction occurs during SARS-CoV infection was not addressed in this paper due to the use of WT SARS-CoV being strictly controlled and the unavailability of a valid anti-P6 antibody.
The interaction between the innate immune response and SARS-CoV has been studied with great interest. IFNs not only modulate SARS-CoV infection of monocytes/macrophages but also affect the severity of the infection. It was found that the elderly are more susceptible to serious infection-related complications, which seem to be related to the fact that IFN-α levels tend to be lower in older people(Wong et al., 2003). Treatment with IFN has been shown to lead to inhibition of SARS-CoV replication in Vero E6 cells(Hensley et al., 2004; Stroher et al., 2004) and decreased expression of SARS-CoV proteins in infected monkeys(Haagmans et al., 2004). However, SARS-CoV has specific mechanisms by which it fights against the host IFN system. In contrast to other coronaviruses, SARS-CoV could prevent IFN-β production upon infection and can suppress IFN immune signaling through directly acting on host proteins via P6(Spiegel et al., 2005). IRF-3, the translocation of which from the cytoplasm to the nucleus is crucial for the activation of antiviral signaling, can be inhibited and detained in the cytoplasm by SARS-CoV P6(Spiegel et al., 2005). Here, we suggest a new potential mechanism by which SARSCoV P6 suppresses the host IFN system by mediating ubiquitination and degradation of the host immune regulator, Nmi. The redundant functions of P6 may maximize its inhibitory role upon the host immune response during SARS-CoV infection. In addition, other studies have provided strong evidence that P6 can enhance SARS-CoV replication as well as its pathogenicity by interacting with host replication and assembly machinery(McBride and Fielding, 2012). Therefore, the performance of rigorous functional studies of SARS-CoV P6 is particularly important for future understanding of the pathogenesis of SARS-CoV and for developing novel anti-virus drugs.
-
This work was supported by China NSFC grants(#31170152 and 81130083). We thank Dr. Stanley Perlman for pCAGGS-P6-HA and pEGFP-P6 constructs.
-
The authors declare that they have no conflict of interest. This article does not contain any studies with human or animal subjects performed by any of the authors.
-
RL carried out the yeast-two hybrid screening assays. SC carried out sequence alignment and molecular cloning, and participated in cell transfection and Western Blot assays. WC carried out cell transfection, Western Blot assays, the immunoprecipitation assays, fluorescence imaging assays, performed the statistical analysis and drafted the manuscript. MW guided the research and completed the manuscript. YC and DG have given advice to this research. All authors read and approved the final manuscript.

 DownLoad:
DownLoad: