










HTML
-
Respiratory syncytial virus (RSV) is the leading viral pathogen associated with bronchiolitis and lower respiratory tract disease in infants and young children worldwide (Psarras et al., 2004). There is a strong association between RSV infection in infancy and the subsequent development of recurrent wheezing and airway hyperresponsiveness later in life (Sigurs et al., 2000). Among adults, the greatest impact of RSV is associated with chronic obstructive pulmonary disease (COPD) (Falsey et al., 2006). Furthermore, numerous studies have indicated that RSV infections can become persistent and latent (Hobson et al., 1997; Matsuse et al., 2000; Riedel et al., 2008). This may contribute to the pathogenesis of chronic wheezing and asthma in children who have experienced acute RSV bronchiolitis and may serve as a reservoir for RSV transmission, which would ostensibly lead to reinfection (Schwarze et al., 2004; Tripp, 2004; Sikkel et al., 2008).
The RSV RNA genome has been characterized from a high percentage of patients with stable COPD, and was associated with elevated markers of inflammation at low copy numbers (Seemungal et al., 2001; Borg et al., 2003). Persistent RSV infection has been established in vitro in several types of cells (Bangham and McMichael, 1986; Valdovinos and Gomez, 2003). These data suggest that although RSV usually causes acute infection, this virus may become latent or persistent both in vivo and in vitro, which suggests the potential for persistent low-grade RSV infection in this population. In the acute infection phase, RSV replication evokes retinoic acid inducible gene-I (RIG-I) to mediate the early antiviral response, and Toll-like receptor 3 (TLR3) to mediate chemokine expression in airway epithelial cells (Le Goffic et al., 2007; Liu et al., 2007; Oshansky et al., 2009). To avoid clearance, the virus adopts certain strategies to subvert the host's antiviral response, including upregulation of suppressor of cytokine signaling (SOCS) proteins, which negatively regulate interferon (IFN)-dependent signaling pathways (Vlotides et al., 2004; Pothlichet et al., 2008; Hashimoto et al., 2009). TLR3 is an important factor for the activation of innate immunity and mediates responses to the synthetic analog of viral double-stranded RNA, polyriboinosinic polyribocytidylic acid (poly I:C), which has been used extensively in experimental studies to mimic viral infections (Stowell et al., 2009; Jeanette et al., 2013). SOCS proteins can be induced directly by TLR3 stimulation, and do not directly interfere with its signaling, but rather avoid overshooting activation by regulating paracrine IFN-β signaling (Dalpke et al., 2008; Prêle et al., 2008). Some latent infectious viruses such as hepatitis C and enterovirus result in persistent infection through the induction of SOCS1 and SOCS3 (Bode et al., 2003; Yasukawa et al., 2003). Therefore, SOCS proteins play an important role in the balanced activation of innate immunity during viral infection.
In present study, we established epithelial cell lines that were persistently infected with RSV (hereafter termed RSV-persistent cultures or cells) and evaluated the putative role of SOCSs in the regulation of the antiviral response and inflammatory status.
-
HEp-2 cells were obtained from the American Type Culture Collection (ATCC, Rockville, MD, USA) and grown as monolayers in minimal essential medium (MEM; Gibco, NY, USA) supplemented with 10% fetal bovine serum (FBS), 100 U/mL penicillin, 50 mg/ mL gentamicin, and 50 ng/mL amphotericin B. For viral purifcation, RSV A2 (ATCC VR-1400) was propagated in HEp-2 cells grown in MEM supplemented with 2% heat-inactivated FBS (MEM-FBS). After the maximum cytopathic effect (CPE) was achieved, the cells were harvested and disrupted by sonication in the same culture medium. Viral purification was performed as previously described (Bangham and McMichael, 1986). Pellets containing the purified virus (pRSV) were resuspended in 10 mmol/L phosphate buffered saline containing 15% sucrose and stored in aliquots at −80 ℃.
-
To establish RSV-persistent cultures, HEp-2 cells grown to 50% confluence were seeded into 100-mm Petri tissue culture dishes and exposed to RSV at a multiplicity of infection (MOI) of 3. The cultures were refreshed with 5% FBS in MEM every three days to remove the floating cells. On day 14 after RSV infection, most of the cells had died, and the few surviving cells remained attached and grew as clonal sheets on day 18. After these surviving cells grew into monolayers, the cells were isolated using clone cylinders (CLS31666-125EA, SigmaAldrich, USA), trypsinized for collection, and then seeded in 96-well plates at a diluted density of one cell per well for clonal selection; this process was repeated three times. Finally, eight RSV-surviving clones (named A-H) were obtained and designated as "passage 0". A frozen stock was then created after three months of sequential passages. The viral titers were determined through a standard methylcellulose plaque assay (Hashimoto et al., 2009). The clone A was used for further studies.
-
The total cellular RNA was extracted and reverse-transcribed using the cloned AMV Reverse Transcriptase (Invitrogen Life Technologies, Carlsbad, CA, USA). The cDNA was PCR-amplifed with primer sets for the RSV N gene (sense, 5′-CAAATACAAAGATGGCTCT-3′, and antisense, 5′-ACATACCTATTAACCCAGT-3′), the human RIG-I gene (sense, 5′-GTGGAATCACGG ATTAGC-3′, and anti-sense, 5′-TTGTCTGGCATC TGGAAC-3′), the human TLR3 gene (sense, 5′-GAT GCTCCGAAGGGTGG-3′, and anti-sense, 5′-AGG GTTTGCGTGTTTCC-3′), and the human GAPDH gene (sense, 5′-TGATGACATCAAGAAGGTGG-3′, and antisense, 5′-TTACTCCTTGGAGGCCATGT-3′). The qPCR reactions were performed using SYBR Green PCR master mix (Applied Biosystems, Foster City, CA, USA) according to the manufacturer's instructions. Duplicate cycle threshold (CT) values were analyzed using the comparative CT (2ΔΔCT) method (Applied Biosystems, Foster City, CA, USA). The relative amount of target mRNA (2ΔΔCT) was obtained by normalizing to the endogenous GAPDH level. The RT-PCR primer sets were as follow: for IFN-β, forward; 5'-AACAAGTGTCTCCTCCAAA-3', reverse; 5'-TCTTCCAGGACTGTCTTCA-3', for 2, 5-OAS1, Forward; 5'-AAGGTGGTAAAGGGTGGCT-3', reverse; 5'-GTGCTTGACTAGGCGGATG-3'; MxA forward 5'-GTTTCCGAAGTGGACATCGCA-3' and reverse 5'-GAAGGGCAACTCCTGACAGT-3'; the thermal cycler conditions were as follows. The reactions were denatured for 2 min at 95 ℃ and then run for 40 cycles of denaturation for 30 s at 95 ℃, annealed for 30 s at 62.5 ℃. The products were separated on 2% agarose gels and visualized through ethidium bromide staining.
-
The cells were fixed, permeabilized in cold acetone, exposed to 5% nonfat milk to block nonspecifc antibody binding, and then incubated with a specific goat anti-RSV F protein polyclonal antibody conjugated to fluorescent isothiocyanate (FITC) at a concentration of 1:500 (Chemicon International Inc., Temecula, CA, USA). The RSV antigen-positive cells (green fluorescence) were counted from fve different randomly selected felds on two slides from each group and observed under fluorescence microscopy (Olympus, Japan).
-
The medium of tenth-generation RSV-persistent cells was refreshed when the cells grew to 60% confluence, and the supernatants were collected 8 h later to determine the IFN-β concentration through ELISA. For acute infection, RSV A2 was inoculated into cells grown to 70% confluence at MOI 3, and the supernatants were collected 8 h later for testing. For poly I:C, HEp-2 cells grown to 70% confluence were pretreated with poly I:C at a fnal concentration of 10 µg/mL, and the supernatants were collected 8 h later for testing. The concentrations of cytokines in duplicate were determined using a commercial sandwich enzyme-linked immunosorbent assay kit (PBL Biomedical Laboratories, Piscataway, NJ, USA) according to the manufacturer's protocol.
-
For siRNA transfection, cationic lipid complexes were prepared according to the instructions for StealthTM RNAi-LipofectamineTM 2000 (Invitrogen Life Technologies, Carlsbad, CA, USA). HEp-2 cells were transfected with 50 nmol/L human siRNA (Invitrogen Co., Shanghai, China) 24 h before RSV infection using StealthTM RNAi-LipofectamineTM 2000. Briefly, 50 nmol/L siRNA was added to a mixture of antibiotic-free MEM-FBS and StealthTM RNAi-LipofectamineTM 2000, and the mixture was incubated for 10 min at room temperature. For transfection, 400 μL of this mixture was added to 24-well plates. The cells were then subjected to qPCR or western blot analysis. Control siRNA was purchased from Invitrogen. The sequences for the human SOCS1 (NM_003745), TLR3 (NM_003265), and RIG-I (NM_014314) siRNAs were as follows: human SOCS1 siRNA, 5′-GCAUCCGCGUGCACUUUCAdTdT-3′; human TLR3, 5′-GCUCGAUCUUUCCUA CAACdTdT-3′; and human RIG-I siRNA, 5′-GCAAGAUCUUACUCA GAGAdTdT-3′. The final concentration of siRNA was 20 nmol/L in all experiments. The transfected cells were placed in a 5% CO2 incubator at 37 ℃.
-
The cells were lysed in buffer containing 20 mmol/ L Tris-HCl (pH 7.4), 0.5% sodium deoxycholate, 10% glycerol, 150 mmol/L NaCl, 2 mmol/L EDTA, 50 mmol/ L glycerophosphate, 2 mmol/L Na3VO4, 10 mmol/L NaF, 1 mmol/L DTT, 1 mmol/L PMSF, and 0.1% protease inhibitor cocktail (Roche, Penzberg, Germany). The protein concentrations were determined using a DC protein assay kit (Bio-Rad, Hercules, CA, USA). The proteins were electrophoresed on SDS polyacrylamide gels and electroblotted onto polyvinyldifluoride membranes (Immobilon, Millipore, Schwalbach, Germany). The membranes were incubated with primary antibodies: goat anti-human SOCS1 pAb, rabbit anti-human SOCS-3 pAb, mouse anti-human STAT1/2 pAb, rabbit anti-human phosphorylated STAT (pSTAT) 1/2 (Santa Cruz, CA, USA), rabbit anti-human RIG-I pAb, rabbit anti-human TLR3 pAb (Cell Signaling Technology, MA, USA), and mouse anti-human β-actin mAbs (Sigma-Aldrich). The membranes were then incubated with horseradish peroxidase-conjugated anti-mouse or anti-rabbit immunoglobulin antibody for 1 h. The antibody-reactive protein bands were visualized by enhanced chemiluminescence (ECL) and western blotting detection reagents (Amersham, Buckinghamshire, UK) and documented with an Image Reader. The density of each band was measured using the NIH image program.
-
The results are expressed as the means ± SE, the signifcance of the data was analyzed using paired Student's t tests; p < 0.05 was considered signifcant.
Virus purifcation and cell cultures
Isolation of RSV-persistent cultures
Reverse transcription polymerase chain reaction (RT-PCR) and real-time quantitative PCR (qPCR) analyses
Immunofluorescence microscopy
Enzyme-linked immunosorbent assay (ELISAs)
Short-interfering RNA (siRNA)-mediated targeted gene silencing
Western blotting
Statistical analysis
-
Using the clone cylinders and three rounds of limited diluted selection, eight RSV-surviving clones (A-H) were obtained, and the RSV N gene was detected in each "passage 0" culture through qPCR. The number of N gene copies of each clone ranged from 2.5 × 102 to 3.5 × 105 copies/mL. During the following months of sub-cloning and passaging, these RSV-surviving clones exhibited two distinct development profiles. Five clones (A, B, E, G, and H) grew slowly with a moderate CPE. In contrast, clones C, D, and F lost their viral infection characteristics; i.e., these clones showed no morphological signs of infection, did not produce detectable viral particles, and did not present any viral antigens and N genes (not detected by immunofluorescence or qPCR) (Figure 1A).
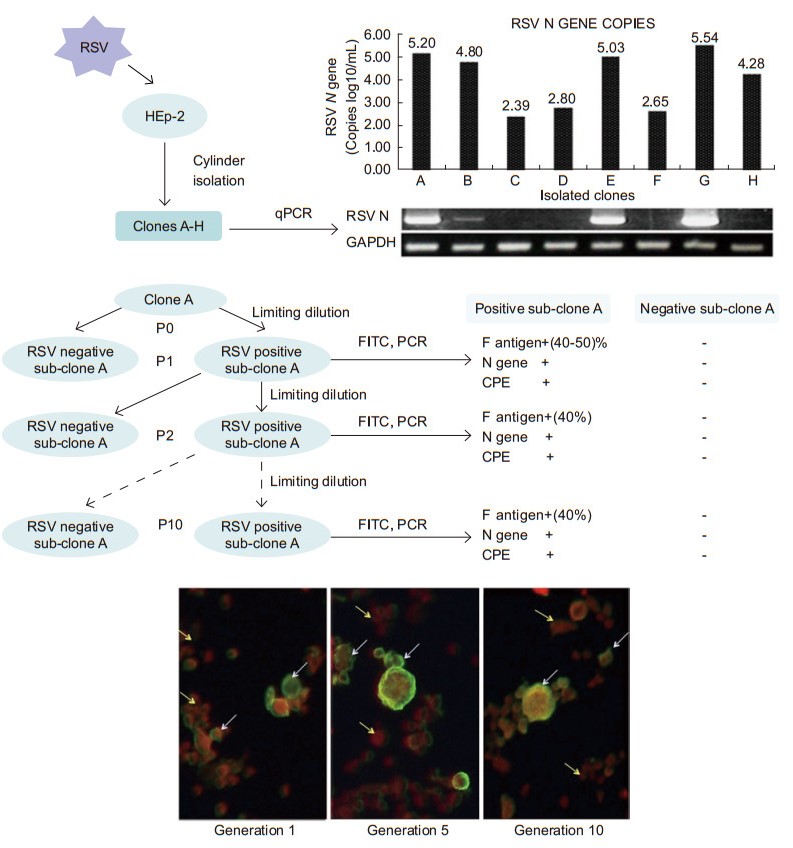
Figure 1. (A) The protocol for the isolation of RSV-persistent cells and quantification of RSV N gene copies. Clones A-H (passage 0) were seeded on 12-well plates at a density of 1 × 105/mL. Seventy-two hours after plating, the total cellular RNA was extracted, and qPCR was performed to quantify the RSV N gene copies. The qPCR products were visualized on 2% agarose gels. (B) Sub-cloning of clone A and characterization. Ten rounds of limiting dilution were performed, and each generation was examined for F antigen, N gene, and CPE. Representations of RSV-persistent cells under fluorescent microscopy. White arrow: RSV positive sub-clone A; yellow arrow: RSV negative sub-clone A.
From the sub-clones with the same characteristics of RSV persistence, clone A was picked up for subsequent analyses, and 10 generations of RSV-positive sub-clones were consecutively isolated by seeding half of the cells in each dilution per well on 96-well plates. After each round of selection, a single cell always grew and differentiated into two distinct states: one that maintained the RSV-persistent characteristics and another that had lost RSV infection characteristics. The RSV-persistent cells grew slowly and formed syncytia. In contrast, neither CPE nor viral antigen could be observed in the clones that lost RSV-infected characteristics. To further confrm that RSV-persistent cells differentiated into two distinct types, the viral antigen (F protein) was detected using an immunofluorescence assay. In each generation of the RSV-persistent sub-clone A, 40%-50% of the cells were RSV F antigen-positive and the remaining cells were RSV antigen-negative (Figure 1B).
The cells with RSV-persistent characteristics exhibited a periodic cell crisis after growing to almost 80% confluence with a severe CPE. Therefore, the RSV-persistent cells were always passaged at 70% confluence to avoid severe CPE. No CPE can be observed in RSV negative sub-clone A (Figure 2A). However, when re-infected with wild-type RSV A2, the RSV-persistent cells were resistant to the re-infection (Figure 2B). These results suggest that the RSV-persistent clone A is heterogeneous with a mixture of variants, and that a single RSVpersistent cell can divide into an RSV-persistent population and an RSV-free population, in which the former is a mixture of RSV antigen-positive and -negative cells, and the latter consists of cells that have lost their RSV infection characteristics.
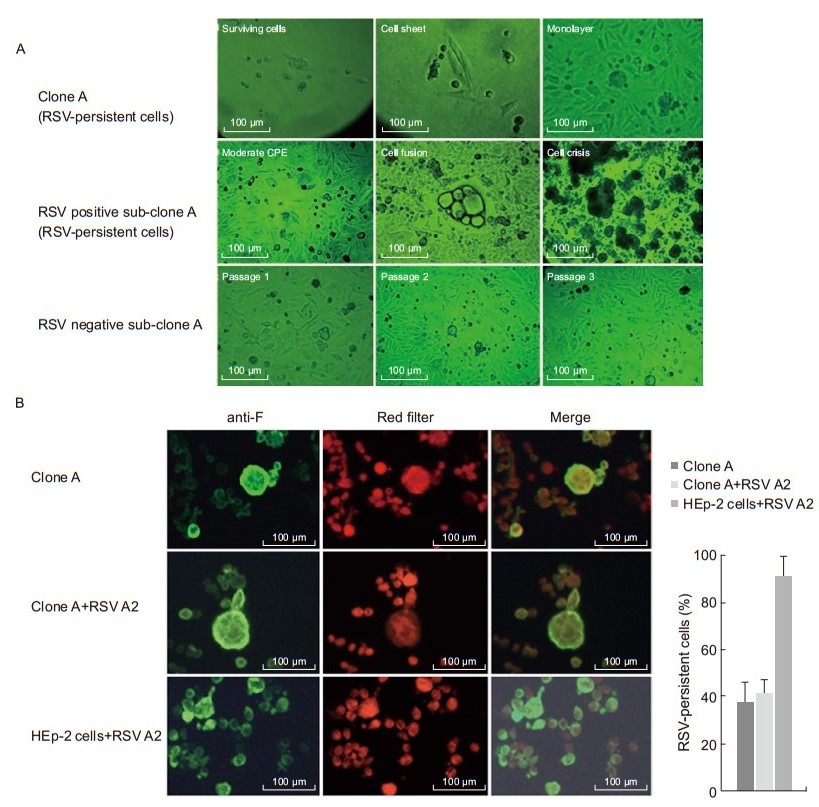
Figure 2. (A) Characteristics of RSV-persistent cells. The top row shows the clone A, at day 14 (surviving cells), day 18 (cell sheet), and day 21 (monolayer) post RSV infection. The middle row shows that the RSV positive sub-clone A (RSV-persistent cells) grew with a moderate CPE, cell fusion at 80% confluence, and cell crisis. The bottom row shows that the RSV negative sub-clone A lost its viral infection characteristics and exhibited "normal" cell morphology during passages 1 to 3, respectively (20× objective, scale bar 100 μm). (B) Detection of RSV F antigens after RSV A2 re-infection. From left to right, RSV F antigen (green), natural light (red flter), and merge, respectively (20× objective, scale bar 100 μm). Quantifcation is shown in the histogram on the right.
-
To analyze RSV propagation and virulence during the persistent state, the tenth generations of the RSV-positive sub-clone A (RSV-persistent cells) and RSV negative subclone A were selected for testing. Cells at 60% confluence were transferred to fresh 2% MEM-FBS medium, and the supernatants of the cultures were collected for plaque assays at various time points. The viral yields of sub-clone A with RSV were relatively low and ranged from 1 × 103 to 1.4 × 104 plaque-forming units (pfu)/mL over the next six days. In contrast, the viral yields of RSV negative sub-clone A reached a peak of 1.5 × 106 pfu/mL on day 3 when re-infected with wild-type RSV A2 at MOI 1 (Figure 3), which is the same result obtained with primary RSV A2 infection in HEp-2 cells. The original HEp-2 cells were then inoculated with the supernatants of the RSV-persistent sub-clone A at MOI 1. CPE was observed at 48 h post-infection, and the viral titer increased to 1 × 106 pfu/mL. When RSV-persistent cells were re-infected with RSV A2 at the same MOI used to establish acute infection, the viral propagation and cell morphology remained unchanged. These results demonstrate that after their development into distinct cell clones, the RSVpersistent cells retained a low rate of replication with the ability to be infective and virulent as well as the ability to resist RSV re-infection, whereas the other type of clone lost its RSV infection ability, became morphologically "normal", and was unable to defend itself against RSV infection.
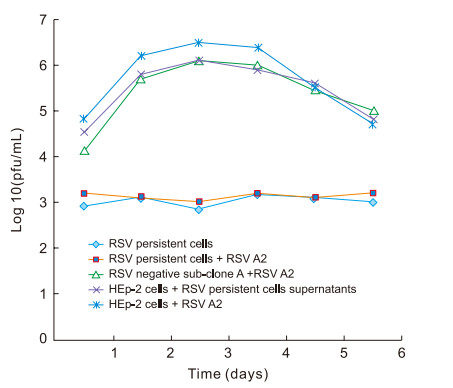
Figure 3. Viral replication in RSV-persistent cultures. The tenth generations of RSV-persistent cells, RSVnegative sub-clone A, and HEp-2 cells were seeded into 12-well plates, and exposed to RSV A2 at a MOI of 1 (re-infection), respectively. In addition, HEp-2 cells were infected with the supernatants of RSV-persistent cells (positive sub-clone A) at MOI 1. Supernatants of all above four situations, together with RSV persistent cells, were collected for a plaque assay at the indicated times.
-
Because viral replication induces antiviral gene expression mainly through the IFN pathway, it could be speculated that this signaling activation is blocked by some negative regulatory mechanism. Therefore, the expression levels of IFN-β were detected using ELISA and the negative regulatory molecules SOCS1 and SOCS3 were detected using western blot.
The RSV-persistent cells were found to secrete more IFN-β (340 ± 110 pg/L) compared to cells with acute RSV infection (160 ± 70 pg/L). Previous studies have confirmed that RSV lytic infection causes transient expression of SOCS1 and SOCS3 within 24 h and that this expression decreases to the basic level after this time period (Hashimoto et al., 2009). In this study, a western blot analysis was performed 8 h after the RSV A2 infection and the medium of the RSV-persistent culture was refreshed. SOCS1 expression significantly increased in the RSV-persistent, acute infection, and poly I:C-treated cells. In contrast, SOCS3 was only weakly expressed in the RSV-persistent cells compared with strong expression observed in the cells with RSV acute infection (Figure 4A). The qPCR results demonstrated that both acute and persistently infected cells displayed up-regulation of either RIG-I or TLR3 mRNA, although a higher level of TLR3 mRNA expression was observed in the RSVpersistent cells (Figure 4B). Viral replication up-regulated the expression of innate immunity-related molecules and activated the IFN-β signaling pathway, as observed in RSV acute infection, and the negative regulatory molecule SOCS1 was also upregulated, which may contribute to impairment of the host's innate antiviral response.
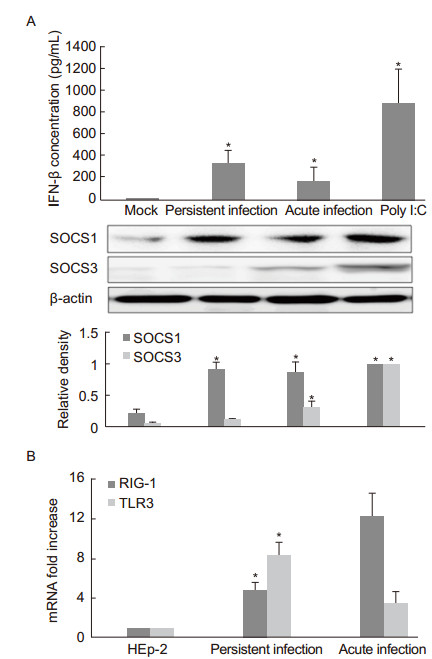
Figure 4. (A) Determination of IFN-β concentration and SOCS1 and SOCS3 expression in RSV-persistent cells. The RSV-persistent cells were seeded on 24-well plates, and the medium was refreshed when cells grew to 70% confluence. After 8 h of culture, duplicate supernatants were collected for the determination of IFN-β concentration by ELISA, and the cellular proteins were harvested for a western blot assay. For RSV acute infection, HEp-2 cells grown to 70% confluence were inoculated with RSV at a MOI of 3 for 8 h, and the supernatants and cellular proteins were collected. HEp-2 cells pretreated with poly I:C (10 μg/mL) for 8 h were used as the IFN-β induction controls. The SOCS1 and SOCS3 protein levels were optimized to a value of 1 relative to the poly I:C-treated cells. Data are shown as the relative density compared to the internal control β-actin of three independent experiments. *p < 0.05 vs. untreated cells (mock). (B) RIG-I and TLR3 mRNA expression in RSV-persistent cells. The medium was refreshed when the cells grew to 70% confluence. The total cellular RNA was extracted for the qPCRassay after 8 h of culture. The data were normalized to GAPDH mRNA levels and are shown as the mean ± SE of three independent experiments. *p < 0.05 vs. acute infection.
-
To clarify whether SOCS1 expression is modulated by RIG-I or TLR3 activation, RNA interference was performed to inhibit the target genes. The tenth generation of RSV-persistent cells were transferred to fresh medium and treated with siRNAs against RIG-I or TLR3 at a fnal concentration of 20 nmol/mL for the indicated times. The cellular extracts were collected for western blot analysis. Inhibition of TLR3 significantly decreased SOCS1 expression at 12 h after siRNA transfection in RSVpersistent cells (Figure 5A), whereas inhibition of RIG-I had no effect on the expression of SOCS1 (Figure 5B). Furthermore, the mRNA levels of IFN-β and its inducible genes MxA and 2, 5-OAS1 were detected using RTPCR. As shown in Figure 5C, the expression of IFN-β, MxA, and 2, 5-OAS1 mRNAs decreased over time when TLR3 was silenced, whereas no change was observed when RIG-I was silenced (Figure 5D). These data indicate that the expression of SOCS1 is related to activation of the TLR3 signaling pathway in RSV-persistent cells, and also contributes to activation of the IFN-β-inducible antiviral response.

Figure 5. Expression of SOCS1 and SOCS3 in RSV-persistent cells. Cells of the RSV-persistent cells were grown to 70% confluence and pretreated with short interfering RNA (siRNA; 20 pmol/mL) for the indicated times; the mismatched siRNA were assigned as controls (mock). The cellular proteins were then harvested for a western blot assay. (A) Expression of SOCS1 in RSV-persistent cells with TLR3 inhibition. (B) Expression of SOCS1 in RSV-persistent cells with RIG-I inhibition. Data are normalized to the internal control β-actin and are shown as the relative density compared to time zero as 1. All data shown are the means ± SE of three independent experiments. *p < 0.05 vs. time zero. Expression of interferon interferon-β (IFN-β) and its inducible genes (MxA, 2, 5-OAS) in RSV-persistent cells (C, D). Cells were pretreated with siRNA and total cellular RNA was extracted for RT-PCR to detect the target genes. The data are representative of one of three identical results.
-
The upregulation of SOCS1 could be related to the balance between innate antiviral immunity and viral replication in RSV-persistent cultures. To clarify the function of SOCS1 on JAK/STAT signaling transduction in RSV-persistent cells, SOCS1 was silenced using siRNA at a fnal concentration of 20 nmol/mL for 24 h, and the cellular extracts were collected to detect the level of STAT1/2 and pSTAT1/2 by western blot. As shown in Figure 6A, the RSV-persistent sub-clone A expressed low levels of phosphorylated STAT1 and STAT2. Silencing of SOCS1 significantly increased the phosphorylation levels of both STAT1 and STAT2. This result implies that upregulation of SOCS1 impairs the innate antiviral response by decreasing pSTAT1 and pSTAT2. Effects of SOCS1 and TLR3 on the expression of chemokines were also observed. RSV-persistent cells were treated with siRNAs against SOCS1 or TLR3 for 24 h, respectively, and the culture supernatants were collected and analyzed using ELISA. No changes in Mip-1α, Rantes, and IL-8 levels were detected in the SOCS1-silenced cells compared to untreated persistent cells, whereas inhibition of TLR3 sharply decreased the secretion of Mip-1α, Rantes, and IL-8 (Figure 6B). Therefore, despite the fact that SOCS1 inhibits endogenous IFN-β signaling but does not affect TLR3 signaling in chemokine activation, RSVpersistent cells avoid the excessive expression of IFN-β and maintain an adaptable antiviral response.
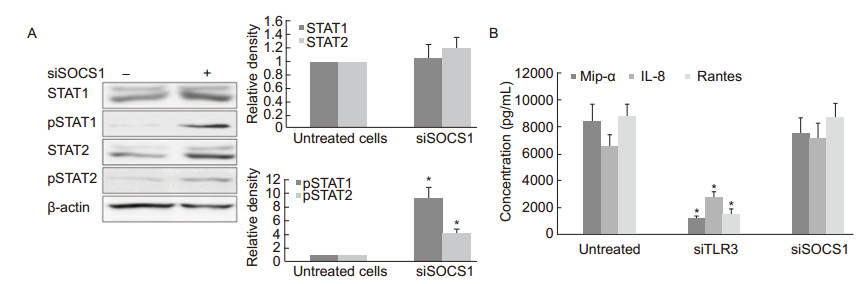
Figure 6. (A) STAT1 and STAT2 phosphorylation in RSV-persistent cells. RSV-persistent cells were grown to 70% confluence and pretreated with siRNA against SOCS1 (siSOCS1) for 24 h. The cellular proteins were harvested for a western blot assay to detect the levels of STAT1, STAT2, pSTAT1, and pSTAT2. Data are normalized to levels of the internal control β-actin and are shown as the relative density compared to untreated cells (designated as 1). (B) Cells were pretreated with siRNA against SOCS1 or TLR3 for 24 h. The duplicate supernatants were subjected to ELISA to determine the chemokine concentrations. *p < 0.05 vs. untreated cells.
Characterization of RSV-persistent cells
Viral replication in RSV-persistent cultures
Antiviral gene expression levels and negative regulation in RSV-persistent cells
Upregulation of SOCS1 is related to TLR3 activation in RSV-persistent cells
SOCS1 modulates STAT1/2 phosphorylation in RSV-persistent cells
-
In this study, we used purified RSV to infect human epithelial cells and established persistent virus-infected clones. The results demonstrate that RSV-persistent cells are heterogeneous and contain different variants: during the passage of cells, half of the cells were viral antigen-positive, and the other half were viral antigen-negative. These RSV-persistent cells secreted high levels of cytokines/chemokines, and showed upregulated expression of SOCS1, which helps the virus to impair the innate antiviral response by inhibiting STAT1/2 phosphorylation.
A cloned single RSV-persistent cell could always develop into a differentiated viral-persistent and "virus-free" culture, but the mechanism of this phenomenon is not clear. One possible mechanism may be the imbalance in the abundance of viral particles and the innate immune status between the two cell types; cells with more viral particles and an impaired innate immune are more prone to be RSV-positive, whereas those with fewer viral particles and innate immune activation are more prone to become RSV-negative. In the heterogeneous persistent culture, the survival of RSV-negative cells depends on the coexistence of RSV-positive cells, because the former cannot resist re-infection alone. This result is consistent with Martinez et al.'s observation that the RSV-persistent culture contains cells with variable morphology (Martínez et al., 2009), and that apoptosis of persistently infected cells is inhibited through induction of the expression of anti-apoptotic genes and inhibition of pro-apoptotic genes (Herranz et al., 2011).
At the early phase of acute RSV infection, a small amount of IFN-β is secreted and there is transient expression of SOCS1, which constitutes a feedback regulation mechanism that inhibits the IFN-β-inducible activation of antiviral gene expression (Zhao et al., 2007; Hashimoto et al., 2009). Unlike RSV acute infection, in this study, both abundant IFN-β secretion and SOCS1 overexpression were consecutively found in RSV-persistent cells. SOCS1 is well known as a strong negative regulatory factor that limits the extent of TLR3 signaling by inhibiting type I IFN signal transduction (Baetz et al., 2004). Furthermore, RSV nonstructural proteins could up-regulate SOCS1 expression independently of the TLR3 and RIG-I signaling pathways, and such upregulation of SOCS1 could be responsible for the inhibition of STAT2 activation (Xu et al., 2014). These studies imply that RSV becomes latent or persistent by subverting the innate antiviral response; however, it is not clear why the persistent viral cells survived under virus replication. One possibility is that the cells developed the ability to resist RSV interference of innate immunity, and thus a balance existed between viral replication and cell proliferation. In RSV-persistent cells, the silencing of TLR3 decreased SOCS1 expression, which suggests that TLR3 contributes to the upregulation of SOCS1. However, the overexpression of SOCS1 did not completely inhibit STAT1/2 phosphorylation, and the IFN-β-stimulated genes MxA and 2, 5-OAS were still expressed. The incomplete block of the effects of IFN-β via SOCS1 may lead to a balance between viral replication and clearance. The RSV-persistent cells are found in an inflammatory state characterized by the secretion of high amounts of the type II cytokines Mip-1α, IL-8, and Rantes, which are related to the TLR3 pathway (Groskreutz et al., 2006). The silencing of TLR3 decreased the secretion of cytokines in persistent cells, which indicates that RSV replication upregulates the expression of cytokines in a TLR3 activation-dependent manner. Furthermore, the overexpression of SOCS1 had no effect on TLR3-induced expression of Mip-1α, IL-8, and Rantes.
Taken together, our results demonstrate that in a persistent culture, mediated by the activation of TLR3, RSV replication induced the production of cytokines and suppressed innate antiviral signaling by upregulation of SOCS1.
-
This work was supported by the National Natural Science Foundation of China (No. 81170005 and No. 30973220).
-
The authors declare that they have no conflict of interest. This article does not contain any studies with human or animal subjects performed by any of the authors.
-
JZ, PY and YT carried out the experiments. PY and DZ performed the statistical analysis. DZ conceived and supervised the study, and worte the manuscript. All authous read and approved the fnal manuscript.

 DownLoad:
DownLoad: