












HTML
-
Coxsackievirus A16 (CVA16) is a positive single stranded RNA virus that belongs to the Picornaviridae family (Mao et al., 2014). It is one of the major causes of hand, foot, and mouth disease (HFMD) (Wei et al., 2014). HFMD is characterized by herpetic lesions on the hands, feet and oral mucosa of children under 5 years old (Mao et al., 2014). It is a serious public health threat to children in Asian-Pacific countries and leads to hundreds of deaths (Chen et al., 2013; Sun et al., 2014). Currently, the treatment and control of HFMD are only symptomatic. There are no effective medications or prophylactic vaccine (Mao et al., 2014). Although an extensive investigation has been performed and there has been progress with vaccines for EV71, no valid CVA16 vaccine is currently available (Ren et al., 2015). Increasing numbers of researches have been done to explore the possible pathogenic mechanisms of CVA16. However, the complete pathogenic mechanisms still remains largely unknown (Zhu et al., 2013; Shi et al., 2015).
Long non-coding RNAs (lncRNAs) are transcripts longer than 200 nucleotides without functional protein-coding capacity (Mattick and Makunin, 2006; Djebali et al., 2012; Fatica and Bozzoni, 2014). The majority of non-coding RNAs are IncRNAs (Djebali et al., 2012; Fatica and Bozzoni, 2014). IncRNAs can be divided into several categories based on their genomic region of origin and relative position to the protein-coding genes in the genome. These categories are as follows: sense, antisense, intronic, intergenic, bidirectional and enhancer (Mercer et al., 2009). IncRNAs are involved in various biological processes by functioning as a cis-tether, in cis-targeting and trans-targeting, as an enhancer, a decoy, a scaffold, an allosteric modification, a co-activator or a co-repressor to modulate gene expression (Lee, 2012). Large-scale studies reported that these RNAs are widely transcribed from the genomes of most complex organisms (Mattick, 2011). It is estimated that approximately 90% of the mammalian genome is transcribed (Bertone et al., 2004; Djebali et al., 2012; Fatica and Bozzoni, 2014). However, messenger RNAs (mRNAs) and microRNAs (miRNAs), which are targeted in previous transcriptional profiling studies, account for approximately 1% of all transcribed species (Kapranov et al., 2007; Djebali et al., 2012). A large percentage of the mammalian genome is transcribed as non-coding RNAs, particularly IncRNAs (Mercer et al., 2009). The discovery of multiple classes of non-coding RNAs, that are pervasively transcribed in mammalian cells and involved in specific biological processes, challenges the traditional view of RNA as an intermediate between gene and protein (Wang and Chang, 2011). The marked differences in the expression patterns and abundances of mRNAs and IncRNAs imply the distinct biological role that IncRNAs may play in physiological and pathophysiological processes (Esteller, 2011).
IncRNAs have recently been associated with virus-host interactions. Josset et al. reported that 5, 329 IncRNAs were differentially expressed after influenza A virus and severe acute respiratory syndrome coronavirus (SARS-CoV) infections (Josset et al., 2014). Other studies showed that IncRNAs 7SL and NEAT1 interfere with the HIV-1 virion package and posttranscriptional expression (Wang et al., 2007; Zhang et al., 2013). These data indicate that multiple steps of the virus infection may be regulated by IncRNAs. However, the detailed mechanisms of how IncRNAs are involved during CVA16 infection remain elusive. In this study, we comprehensively characterize the land scape of IncRNAs expressed with or without CVA16 infection in RD cells. Gene Ontology (GO) (Blake and Harris, 2008) and Kyoto Encyclopedia of Genes and Genomes (KEGG) (Du et al., 2014) analyses were adopted to predict the possible physiological activities and related signal pathways. Thus, we reported a comprehensive catalog of IncRNAs and provided a transcriptome blueprint to identify novel molecular targets and pathways for the treatment of HFMD.
-
RD cells were purchased from the American Type Culture Collection (ATCC) and maintained in minimum essential medium (MEM) supplemented with 10% fetal bovine serum (FBS) (SV30087; HyClone) or 2% FBS (maintenance medium). The cells were cultured at 37 ℃ in a humidified incubator with 5% CO2. CVA16 is a laboratory strain that has been completely sequenced and belongs to the B1 genotype (Shi et al., 2015).
-
The total RNA was extracted using TRIzol reagent (Invitrogen, California, USA) according to the manufacturer's guidelines. Total RNA (2 μg) was reverse transcribed into 20 μL cDNA using a Reverse Transcription kit (Thermo, Massachusetts, USA) according to the manufacturer's protocol. The cDNA samples were subjected to real-time PCR (Bio-Rad iQ5; Bio-Rad, California, USA) using SYBR green (Gene Copoeia Inc., Maryl and, USA) and GAPDH as an internal control. The primers are listed in Table 1. To determine the IncRNAs levels, the following conditions were used: 94 ℃ for 5 min, followed by 40 cycles of 94 ℃ for 10 s, 57–65 ℃ for 20 s and 72 ℃ for 30 s. All samples were run in triplicate, and the data analysis was performed using the 2–ΔΔCt method.
-
RD cells were infected with CVA16 until cytopathic effects were observed. Total RNA was isolated from each sample by using TRIzol reagent (Invitrogen) according to the manufacturer's protocol. The RNA concentration of each sample was determined by measuring the absorption at 260 and 280 nm using Nanodrop (Genergy Co., Shanghai, China). High-throughput RNA-Seq of the two mixed samples was performed on Illumina Hiseq2500 with a 101 bp double-end protocol (Genergy Co., Shanghai, China) (Trapnell et al., 2010). Given that the poor-quality fractions of the sequencing data were highly distributed in the end and Trim Galore software was used to dynamically remove the poor-quality segments (Wang et al., 2012). Then, FastQC software was adopted for quality control analysis. Subsequently, TopHat was used to map the pre-proposed reads to Homo_sapiens GRCh37 and analyze the mapping results to identify splice junctions between the exons (Trapnell et al., 2009). The mapping reads were arranged using Cufflinks to assemble transcripts and estimate their abundance (Borodina et al., 2011). For IncRNAs analyses, the Ensembl, Gencode, NCBI RefGene, UCSC lincRNA, Lncipedia and Noncode databases were chosen as annotation references. For mRNA analyses, we adopted RefSeq and Ensembl databases. Candidate IncRNAs were acquired by satisfying the following criteria: RNA length 200 nt, CPC score 0, CPAT probability 0.364 and phyloCSF score –20. The expression levels of the transcripts were calculated by fragments per kilobase of transcript per million fragments mapped (FPKM) values. Differentially expressed transcripts (DETs) were defined as P < 0.05 and/or fold change > 2 times based on their FPKM values between the groups, which were identified using Cuffdiff software (Trapnell et al., 2010).
-
Secondary structure analysis was performed with RNAfold (Vienna package, http://rna.tbi.univie.ac.at/cgi-bin/RNAfold.cgi). The structure is a minimal free energy structure and base pairing probabilities have been color coded using a scale from 0 (blue) to 1 (red).
-
Gene ontology (GO) was adopted to annotate the functions of differentially expressed genes in the GO vocabularies (Blake and Harris, 2008). Briefly, the differentially expressed genes were regarded as candidates from the whole genes and the differentially expressed genes were calculated using a hypergeometric distribution test. The P-value was further corrected by Benjamini-Hochberg multiple test to obtain the false discovery rate (FDR) and based on P-value and FDR, the enrichment score was expressed in -log 10 (P-value).
Kyoto Encyclopedia of Genes and Genomes (KEGG) pathway analysis was also used to define the functions of the differentially expressed genes in graphical diagrams of biochemical pathways (Du et al., 2014). KEGG pathway analysis was similar to that of GO functional analysis. The significance was calculated by FDR and P-value.
-
IncRNAs can regulate the expressions of genes that are located on the same chromosome, and such regulation is called cis regulation (Huang et al., 2012; Kornienko et al., 2013). Several classes of IncRNAs have been reported to regulate their protein-coding host genes in cis manners such as sense intronic, natural antisense transcripts, bidirectional and long intergenic non-coding RNA (Huang et al., 2012; Kornienko et al., 2013). In the present study, we subjected the differentially expressed known IncRNAs to cis analysis by built-in perl code program.
IncRNAs also can act in a trans manner when they affect genes on other chromosomes (Gong and Maquat, 2011; Huang et al., 2012). Previous studies have shown that IncRNAs can interact with the 3'untranslated region (3'UTR) of its co-expression mRNA, forming complementary hybrids (Gong and Maquat, 2011). Therefore, using the RNAplex and Risearch program (Tafer and Hofacker, 2008; Wenzel et al., 2012), we subjected the differentially expressed known IncRNAs to trans analysis.
Cells and viruses
RNA extraction and real-time PCR
RNA-seq and analysis
Secondary structure prediction
GO and KEGG pathway enrichment analyses
Cis and trans analyses
-
To investigate the roles of host IncRNAs in CVA16 infection, genome-wide RNA-seq was performed using human RD cells infected with or without CVA16. A total of 204 million reads were obtained. The control sample resulted in 95 million reads, and the virus-infected sample resulted in 109 million reads. We mapped RNA-seq reads to the human reference genome using TopHat. Transcripts were reconstructed using Cufflinks. HMMer+Pfam, CPC, PhyloCSF and CPAT were used to calculate the coding potential of transcripts. The transcripts that passed HMMer+Pfam, CPC, PhyloCSF and CPAT coding potential filters were further selected as potential IncRNAs. Our stringent strategy is summarized in Figure 1A. Our transcriptome contained a total of 1, 970 IncRNAs and 6, 416 mRNAs with differential expression. Using the criteria of a corrected log2 (fold_change) 2, we identified 760 upregulated and 1, 210 downregulated IncRNAs (Figure 1B, Supplementary Table S1). These IncRNAs were significantly and differentially expressed in CVA16 infected cells compared to the control. Out of the 6, 416 mRNAs, 3, 861 were upregulated and 2, 555 downregulated (Supplementary Table S2). Next, we explored the relative distribution of expressed IncRNAs and mRNAs derived from each chromosome. We found that these reads are ubiquitously distributed in all chromosomes including sex and mitochondria chromosomes (Figure 1C, 1D) (Yang et al., 2014). In addition, the expression levels of IncRNAs were markedly lower than mRNAs, which is consistent with previous reports (Dinger et al., 2008).
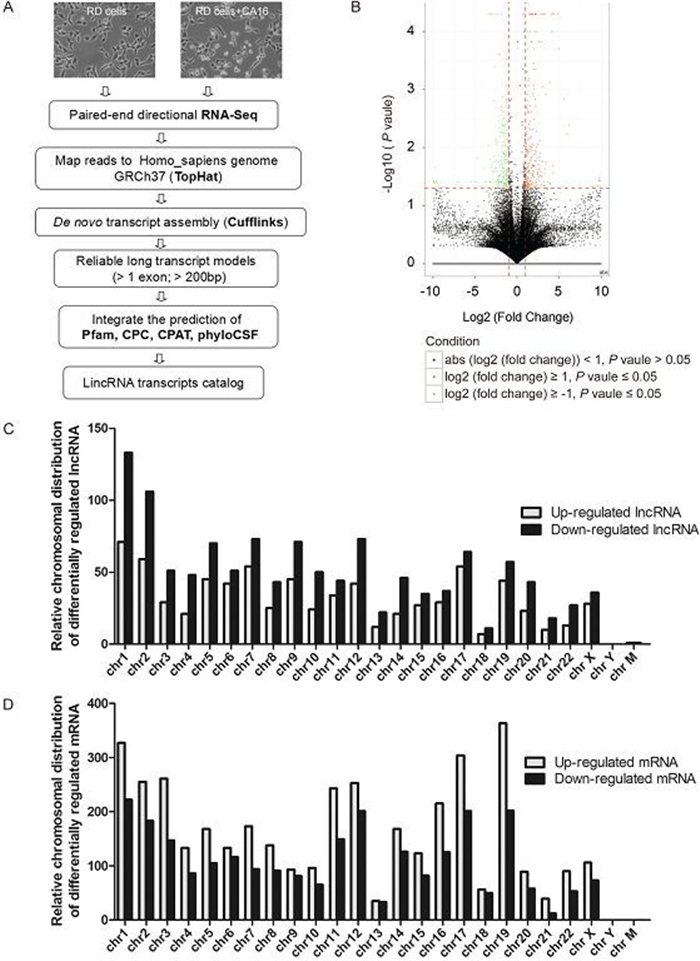
Figure 1. The global discovery of IncRNAs and mRNA expressed in control and CVA16 infected RD cells. (A) Workflow for IncRNAs discovery. The detailed process can be found in the materials and methods. (B) Volcano plots of IncRNAs with significant differential expression. X-axis: log2 fold change; Y-axis: – log10 (FDR P-value); Vertical dotted lines: 2 fold changes in expression level; Horizontal dotted line: the significance cutoff. (C–D) Relative distribution of expressed IncRNAs and mRNA derived from each chromosome.
-
We summarized the general characteristics of dysregulated IncRNAs, such as length distribution, exon number and classification. The majority of the IncRNAs consist of a few exons (less than 5 exons) (Figure 2A). Most IncRNAs are short, with a majority less than 4 kb in length (Figure 2B).
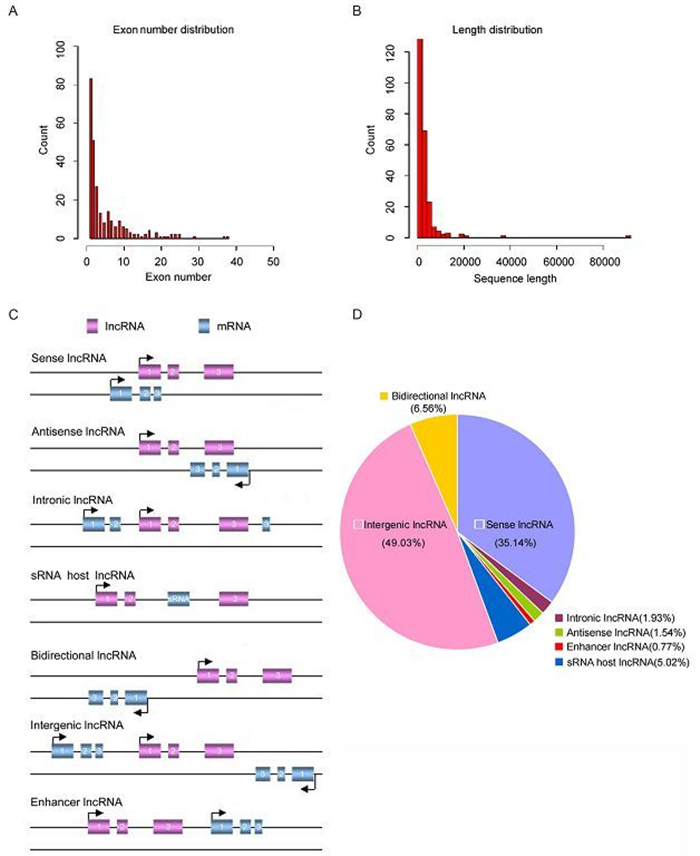
Figure 2. The properties of novel IncRNAs. (A) Number of exons. (B) Size distribution of IncRNAs. (C) Definitions of different classes of IncRNAs based on their genomic region. (D) Annotation of the genomic context of differentially expressed IncRNAs.
Based on their genomic locations relative to adjacent protein-coding genes, IncRNAs can be divided into several categories: sense, antisense, intergenic, intronic IncRNAs, enhancer IncRNAs or bidirectional IncRNAs (Figure 2C). In the current study, we classified the dysregulated IncRNAs and discovered that 43.64% were intergenic, 22.31% were sense, 15.89% were intronic, 8.67% were bidirectional, 5.59% were antisense, 3.85% were sRNA host IncRNAs and 0.05% were enhancer (Figure 2D). Among the successfully annotated IncRNAs, more intergenic transcripts than antisense or intronic transcripts were differentially expressed, which were consistent with previous other investigations (Chen et al., 2015; Yu et al., 2015).
-
In this study, to further validate the accuracy of IncRNAs profile determined by RNA-seq, six IncRNAs which are less than 10 kb were selected as candidates and confirmed by semi-quantitative RT-PCR (Figure 3A, 3B) and quantitative real-time PCR (Figure 3C). As shown in Figure 3C, the real-time PCR results indicate that in the CVA16 infected RD cells the expression levels of NONHSAT102806 and ENST20160205307533 increased 2 fold and 4 fold respectively. Meanwhile, the RNA-seq data reveal that the fold change of NONHSAT102806 is 2.6 and ENST20160205307533 is 3.2 upon CVA16 infection (Figure 3D). Next, we compared the real-time PCR results of down-regulated IncRNAs with the RNASeq results and discovered that the changed trends and fold change of real-time PCR results are also consistent with the magnitude of RNA-seq (Figure 3B, 3D). Collectively, these data demonstrated that the real-time PCR results of up/down-regulated IncRNAs are consistent with the levels of differential expression.
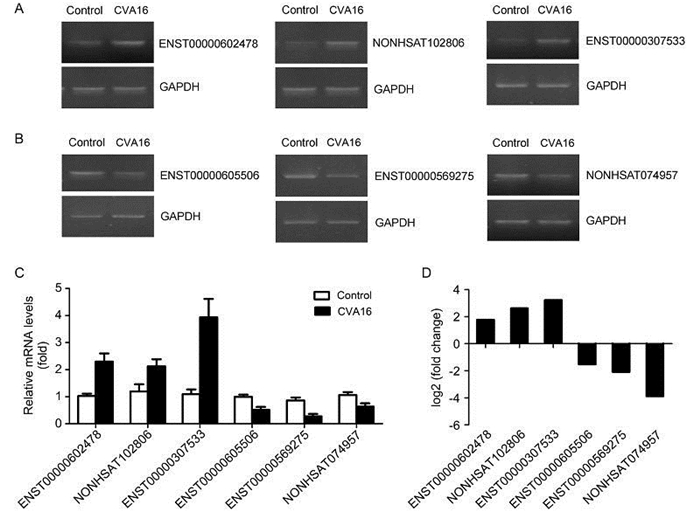
Figure 3. The expression of 6 differentially expressed IncRNAs was validated by semi-quantitative RT-PCR and quantitative real-time PCR. (A–B) Three upregulated IncRNAs and three downregulated IncRNAs, with meaningful elevation or reduction in expression fold demonstrated by RNA-seq assays, were further analyzed and confirmed using semi-quantitative RT-PCR (gel image). (C) Quantitative real-time PCR (column graph) results. (D) RNA-seq results of 6 selected IncRNAs.
-
The GO project provided a powerful tool to construct and use ontologies to facilitate the biologically meaningful annotation of genes and their products in a wide variety of organisms (Blake et al., 2008). Genes differentially expressed upon CVA16 infection were analyzed for GO enrichment using the GOseq software package and the top GO terms (based on P value) for cellular component, molecular function, and biologic process for differentially expressed genes are shown in Figure 4A.
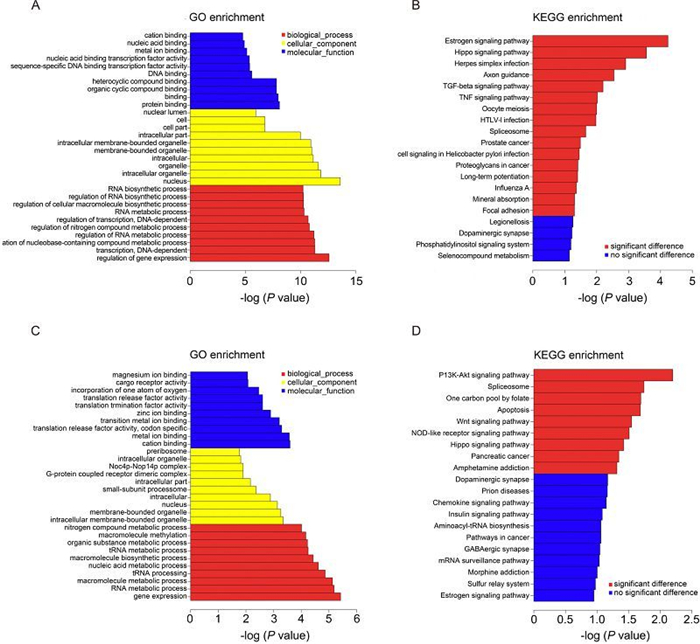
Figure 4. GO and KEGG pathway analysis. (A) The top 10 GO terms of the differentially expressed genes are listed. (B) The pathways that associated with the differentially expressed genes are listed. (C) The top 10 GO terms of the genes that are targeted by differentially expressed IncRNAs. (D) The pathways that were associated with the genes that are targeted by differentially expressed IncRNAs are listed.
For cellular component terms, genes differentially expressed are enriched for components of the nucleus, membrane-bounded organelle, intracellular membrane-bounded organelle and intracellular part (Figure 4A).
For molecular function terms, differentially expressed genes show enrichment for transcripts that encode nucleic acid binding transcription factor activity, sequence-specific DNA binding transcription factor activity. Transcripts encoding binding activities, include protein binding, DNA binding, metal ion binding, nucleic acid binding, heterocyclic compound binding and organic cyclic compound binding are also abundantly expressed in CVA16 infected RD cell fraction (Figure 4A).
Biologic process terms enriched in differential expressed genes include regulation of gene expression, transcription, regulation of RNA metabolic process and regulation of transcription (Figure 4A). This is consistent with the fact that RD cells undergo changes of gene expression regulation after CVA16 infection (Figure 4A).
Next, these genes were subjected to KEGG database analysis to predict the biological pathways associated with the differentially expressed genes (Du et al., 2014). The top 16 significant pathways associated with the differentially expressed genes were shown (Figure 4B). The results of KEGG analysis showed that they were mainly enriched for TGF-beta signaling pathway, TNF signaling pathway and virus infection related signaling pathway (influenza A, HTLV-1 infection).
Target genes of differentially expressed IncRNAs were predicted and these target genes also were subjected to GO annotation and KEGG pathway analysis (Figure 4C, 4D). The results of GO annotation demonstrated the main functions of these target genes were associated with magnesium ion binding, cargo receptor activity, nitrogen compound metabolic processes, molecular methylation and organic substance metabolic processes (Figure 4C). While the results of KEGG analysis showed they were mainly related to the following signal pathways: (1) the PI3K-Akt signaling pathway, (2) the splicesome, (3) apoptosis, (4) the Wnt signaling pathway, (5) the NOD-like receptor signaling pathway, (6) the Hippo signaling pathway and (7) pancreatic cancer (Figure 4D).
-
IncRNAs can work in cis or trans manner when they affect genes on the same or different chromosomes (Gong and Maquat, 2011; Huang et al., 2012; Kornienko et al., 2013). Identification of the genes associated with differentially expressed IncRNAs via cis-or trans-regulation might provide insight into the potential functions of IncRNAs. In the present study, we subjected the differentially expressed IncRNAs to cis analysis, and we found that hundreds of the differentially expressed known IncRNAs could act in a cis manner. As shown in Supplementary Figure S1A, we know that lnc-TENC1-1:1 was associated with eukaryotic translation initiation factor 4B (eIF4B) which is an important protein involved in the initiation phase of eukaryotic translation (Supplementary Table S3). Meanwhile, mitogen-activated protein kinase 9 (MAPK9) are predicted to be associated with UCSC_TCONS_00010210 (Supplementary Table S3).
In addition, we also subjected the differentially expressed IncRNAs to trans -analysis and found that trans-acting IncRNAs may not be as prevalent as cis-acting IncRNAs. In addition, we further detected the networks formed by the trans-acting IncRNAs and their associated genes and found that one IncRNAs can have one or more associated genes. As shown in Supplementary Figure S1B, NONHSAG051892 not only was predicted to be associated with Ribonuclease P protein subunit p38 but also Galactokinase in trans manner. More detailed information can be found in Supplementary Table S3 and Supplementary Table S4.
-
The function of an IncRNAs cannot be inferred from the sequence or primary structure alone (Mercer et al., 2009; Wang and Chang, 2011; Rybarczyk et al., 2015). Indeed, there is already strong evidence that evolutionarily conserved RNA secondary structures are a robust indicator of molecular function. Based on the predicted secondary structure of IncRNAs NRAV, Jing Ouyang et al. conducted a series of related experiments and demonstrated that a small arm of NRAV (nt 618–872) was non-essential for its role in controlling IAV replication (Ouyang et al., 2014). In the present study, we also predicted the second structure of six IncRNAs and we discovered various pseudoknots or stem loops in these IncRNAs (Supplementary Figure S2). It is attempting to estimate that the flexible and complex structures of the RNA may be related to the diversity of their functions and whether these stem loops play vital roles need to be validated further.
To characterize the distinctive chromatin structures, histone modifications and transcription factor binding signatures, we selected ENST20160205602478 as an example for further research. We found that ENST20160205602478 is a natural antisense IncRNAs that is mapped on chromosome 22 (Figure 5A). Two protein coding genes on the opposite strand transcribe in the opposite direction (Figure 5A). One is Cytochrome-b5 reductase (methemoglobin reductase), an NADH-dependent enzyme that converts methemoglobin to hemoglobin. The other protein is the polymerase delta-interacting protein 3, a protein that interacts with the DNA polymerase delta p50 subunit and is also a specific target of S6 kinase 1 (regulates cell growth). Exon of ENST20160205602478 shows high conservation across mammalian species (Figure 5B). Further, there is a prominent histone H3-lysine-27 acetylation (H3K27ac) marker near exon of ENST20160205602478, suggesting the presence of an active promoter (Figure 5B). Further experiments, such as chromatin immunoprecipitation sequencing (ChIP-seq) or multisite ChIP-quantitative polymerase chain reaction (qPCR), are necessary to verify the histone modifications near the IncRNAs loci. Another track DNase Ⅰ hypersensitivity peak clusters also are enriched in transcription start sites (TSS) of ENST20160205602478 (Figure 5B), leading to increased chromatin accessibility. In addition, in the IncRNAs promoter region, we report various binding motifs (+1000 to 0 nt relative to transcription start sites (TSS)) for transcription factors which may regulate IncRNAs expression (Figure 5C). And the critical roles for these transcription factors need to be confirmed by luciferase assays. Collectively, these features indicated that the expression of IncRNAs can be regulated by various mechanisms.
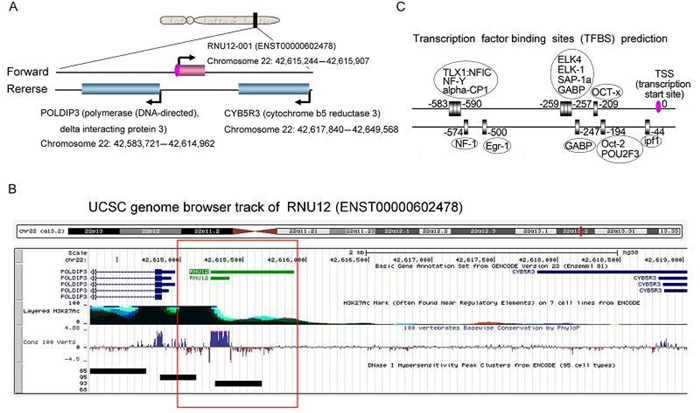
Figure 5. A bioinformatics analysis of ENST20160205602478. (A) The position of ENST20160205602478 and neighboring genes in chromosome 22. (B) Several integrated regulation tracks spanning the ENST20160205602478, including conservation, histone markings and DNase Ⅰ hypersensitivity, are displayed. (C) Transcription factor binding sites (TFBS) that are located in the IncRNAs promoter region (–1000 to 0 nt relative to TSS) were predicted by MotifMap software. The transcription factors (TF) were in the oval boxes and the numbers indicated the binding sites.
The global discovery of IncRNAs expressed in control and CVA16 infected RD cells
General characteristics of dysregulated IncRNAs
Quantitative assays of dysregulated IncRNAs after CVA16 infection
KEGG-pathway analysis
Cis-acting and trans-acting regulatory network
Further analysis about selected IncRNAs
-
The present study utilized next-generation sequencing to provide a quantitative and comprehensive analysis of the coding and non-coding transcriptome of mock-infected and CVA16 infected RD cells. These analyses revealed significant differences in the patterns of mRNA and IncRNAs expression. A total of 1, 970 IncRNAs and 6, 416 mRNAs were identified as significantly and differentially expressed between control and CVA16 infected cells. GO analysis and KEGG analysis were conducted to investigate the potential functions of these IncRNAs. Our study is the first to use comprehensive deep-sequencing technology to clearly demonstrate that IncRNAs are involved in the host response to CVA16 infection.
A number of recent studies have suggested that noncoding RNAs (ncRNAs) function in pathogen-host interactions (Peng et al., 2010; Winterling et al., 2014). Using next-generation sequencing, Peng et al. reported the differential expression of approximately 500 annotated and 1, 000 non-annotated genomic regions during SARS-CoV infection across four founder mouse strains (Peng et al., 2010). Using two commercially available microarray systems, Winterling et al. identified the differential expression of 42 ncRNAs during influenza A virus (IAV) infection in human lung epithelial cells and IncRNAs VIN can facilitate influenza A virus (IAV) propagation (Winterling et al., 2014). Despite these progresses, the specific functions of these IncRNAs remain incompletely characterized and these virus infection models provide a unique platform for studying the biology and regulation of IncRNAs.
Unlike mRNAs, IncRNAs possess some unique properties. In this study, we revealed that the expression levels of most IncRNAs are lower than mRNA. We also noted that more intergenic IncRNAs than antisense and intronic IncRNAs were differentially expressed. This result may be an intrinsic property of organism system associated with the respective functions of non-coding genes and protein-coding genes. Another interesting finding was that the numbers of down-regulated IncRNAs are unexceptionally more than that of up-regulated IncRNAs in each chromosome, and the result of mRNA is quite the opposite. Although this result is consistent with previous other investigations (Bu et al., 2012; Zhu et al., 2015), Yu et al. reported that there were more down-regulated IncRNAs than up-regulated IncRNAs in the glucocorticoids-treated bone microvascular endothelial cells and conversely, differentially up-regulated mRNAs were more common than significantly down-regulated mRNAs (Yu et al., 2015). The precise regulatory mechanism remains unclear and further studies are needed to illuminate the exact mechanisms. Taken together, these results demonstrate that our IncRNAs share similar features with those described in previous studies, in terms of structural, expression, and conservation properties (Alvarez-Dominguez et al., 2014; Yang et al., 2014; Chen et al., 2015).
Although the development of high throughput deep sequencing technology provided the possibility of a nearly complete view of IncRNAs profiles, the identification of IncRNAs function still remains challenging (Wang and Chang, 2011; Djebali et al., 2012). The GO project and KEGG Pathway analysis are widely used to illustrate the differentially expressed genes in terms of functions and pathways. In this study, we detected that the dysregulated genes were enriched in RNA biologic processes including RNA biosynthetic processes, RNA metabolic processes and the regulation of RNA upon CVA16 infection (Figure 4A). The coding gene targeted by dysregulated IncRNAs shown enrichment in the PI3K-Akt signaling pathway, splicesome, apoptosis, Wnt signaling pathway and the NOD-like receptor signaling pathway (Figure 4D). The PI3K-Akt signaling pathway regulates many cellular processes including development, cell proliferation, differentiation, and apoptosis. The NOD-like receptor signaling pathway plays key roles in the regulation of the innate immune response by cooperating with Toll-like receptors and regulating inflammation. Therefore, the GO project and KEGG Pathway analysis provide guidance for further efficient identification of potential functions and regulatory mechanism of IncRNAs. And further experimental verifications are needed to verify the hypothesis.
IncRNAs can regulate gene expression either in a cis (on neighboring genes) or in a trans (on distantly located genes) manner (Mercer et al., 2009; Wang and Chang, 2011; Guil and Esteller, 2012; Lee, 2012). Although an increasing number of IncRNAs are reported, only a small fraction of them have functional annotations. Most IncRNAs in a given cellular content remain enigmas. In the current study, we analyzed IncRNAs-mRNA genomic proximity information and constructed a co-expression network to explore the potential cis-and trans-regulatory roles of IncRNAs on coding mRNAs. We discovered that numerous IncRNAs interacted with associated protein-coding genes in cis or trans manners, which suggests that these IncRNAs might be biologically meaningful. Notably, these IncRNAs were found to form a “many-to-many†network with their associated genes, which reflects the complexity of the mechanisms of the regulation of CVA16-regulated IncRNAs. The IncRNAs and mRNA co-expression networks provide concrete targets for further function research of IncRNAs. Further experiments are needed to investigate the precise natures of these IncRNAs.
These studies utilized next-generation sequencing technologies for comprehensive mRNA and IncRNAs expression profiling in control and CVA16 infected RD cells. These experiments revealed a distinct relative abundance, expression pattern and genomic origin of mRNA and IncRNAs in human RD cells, highlighting the different biological roles of the individual RNA classes. Further study of the nature and function of these dysregulated IncRNAs is necessary to determine the mechanisms of HFMD. All in all we identified a panel of IncRNAs derived from CVA16 infected RD cells, which may provide new targets for the diagnosis, treatment and prevention of HFMD.
-
This work was supported by the National Natural Sciences Foundation of China (No. 81171577, 81371790, 81371422 and 81171127), Major AIDS and Viral Hepatitis and Other Major Infectious Disease Prevention and Control project of China (2014ZX10001003), the Fundamental Research Funds for the Central Universities of China and the Translational Medical Research Fund of Wuhan University School of Medicine.
-
The authors declare that they have no conflict of interest. This article does not contain any studies with human or animal subjects performed by any of the authors.
-
WHL and YYS designed the experiments, analyzed the data and wrote the paper. YYS and HLT performed the majority of the experiments. XC, YYZ, LJC and ZCL offered some experimental materials. JQS, SH, JY, BWP, XHH and WHL supervised this study and reviewed and edited the paper.
-
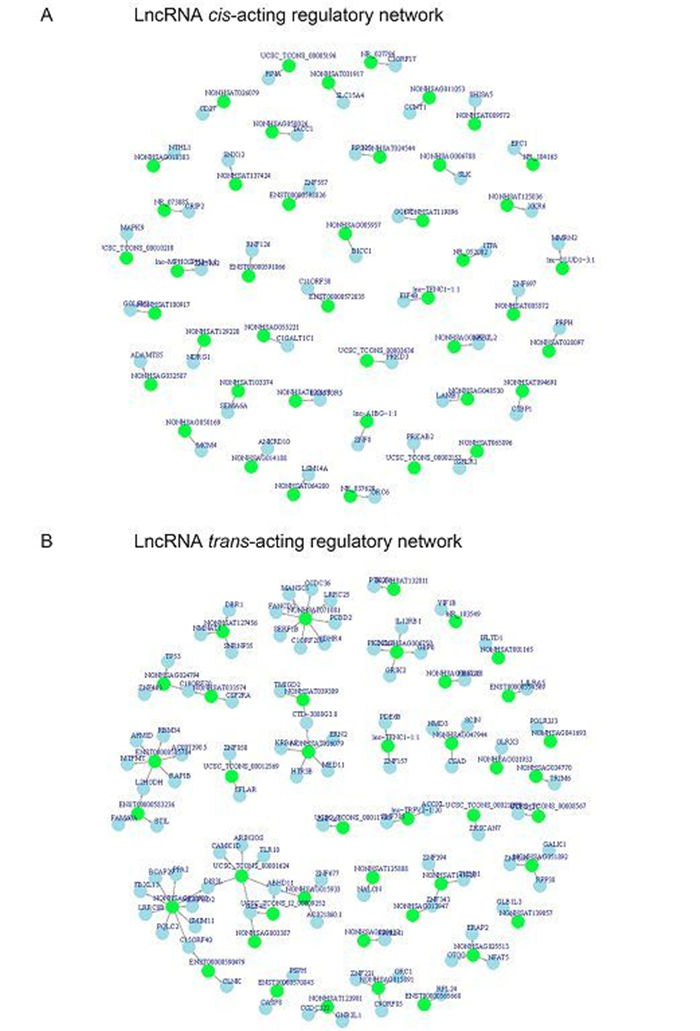
Figure S1. Cis-acting and trans-acting regulatory network. (A) Cis-acting regulatory network; (B) Trans-acting regulatory network. Red circles represent IncRNAs and blue circles represent mRNA.
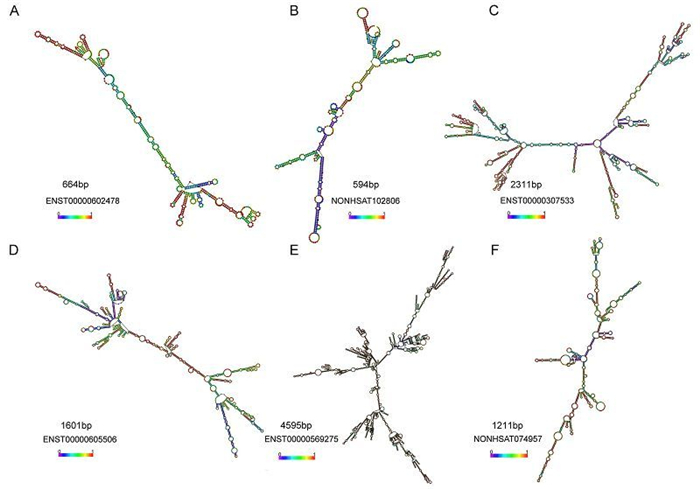
Figure S2. The secondary structures prediction of IncRNAs were performed using RNAfold software. (A–C) RNA secondary structure of three upregulated IncRNAs. (D–F) RNA secondary structure of three downregulated IncRNAs. The second structures are minimal free energy structures. Base pairing probabilities have been color coded using a scale from 0 (blue) to 1 (red).
Table S1. The 1970 differentially expressed lncRNAs expressed in RD cells after CVA16 infection
Table S2. The 6416 differentially expressed mRNAs expressed in RD cells after CVA16 infection
Table S3. Cis-acting lncRNA-mRNA co-expression network
Table S4. Tran-acting lncRNA-mRNA co-expression network

 DownLoad:
DownLoad: