












HTML
-
Coxsackievirus A16(CA16) is one of the major viral pathogens associated with hand, foot, and mouth disease. CA16 belongs to the Enterovirus genus of the Picornaviridae family and possesses a single-stranded positive-sense RNA genome (Mao et al., 2014). Reverse genetics is an important tool for CA16 research. Previously, a reverse genetics T7 polymerase-based system was developed for poliovirus (Moss et al., 1989), foot-and-mouth disease virus (FMDV) (Zibert et al., 1990), coxsackievirus B3(Klump et al., 1990), enterovirus 71(EV71) (Han et al., 2010; Shang et al., 2013), and even CA16(Liu et al., 2011). However, in that system, cDNA must be transcribed to RNA by an exogenous T7 polymerase in vitro or in vivo. To develop a more rapid and simple method, a reverse genetics system based on human polymerase Ⅰ (Pol Ⅰ) was developed for FMDV (Chang et al., 2009) and EV71(Meng et al., 2012). In the current study, we developed a high-efficiency Pol Ⅰ-driven plasmid-based reverse genetics system for CA16(GenBank: EU262658), and systemically characterized recovered CA16 particles.
Construction of pMD19T-pPol Ⅰ-CA16 is shown in Figure 1A. Pol Ⅰ promoter (containing multiple cloning sites [MCS]) was amplified using primers P1 and P2(all primers are listed in Supplementary Table S1) from pHW2000(Hoffmann et al., 2000). It was then inserted into the pMD19T vector (Takara), and the construct was termed pMD19T-pPol Ⅰ-MCS. Two complementary oligo-nucleotides, P3 and P4, containing the murine termi-nator sequence, were designed to anneal and create 5′ and 3′ Not Ⅰ overhangs. After annealing, the oligonucleotides were ligated with Not Ⅰ-digested pMD19T-pPol Ⅰ-MCS. The resultant vector was named pMD19T-pPol Ⅰ-MCS-Ter with only one Not Ⅰ site, because the Not Ⅰ site of 3′murine terminator was disrupted in the preceding step. CA16(1-4388 bp), amplified from pMD19T-CV (Liu et al., 2011) with primers P5 and P6, was inserted into pMD19T-pPol Ⅰ-MCS-Ter Hind Ⅲ and Xba Ⅰ sites, ge-nerating pMD19T-pPol Ⅰ-CA16(1-4388). Next, CA16(4386-7435 bp) amplified from pMD19T-CV using P7 and P8, was inserted into the Xba Ⅰ site of pMD19T-pPol Ⅰ-CA16(1-4388). After confirmation of insert directio-nality, the obtained plasmid was termed pMD19T-pPol Ⅰ-CA16. The construct size was 10, 392 bp, including 7, 435 bp CA16 viral DNA. CA16 genome was thus placed under control of the human Pol Ⅰ promoter, to be transcribed after transfection of mammalian cells. The CA16 sequence in this plasmid was identical to our previously reported sequence (Liu et al., 2011).
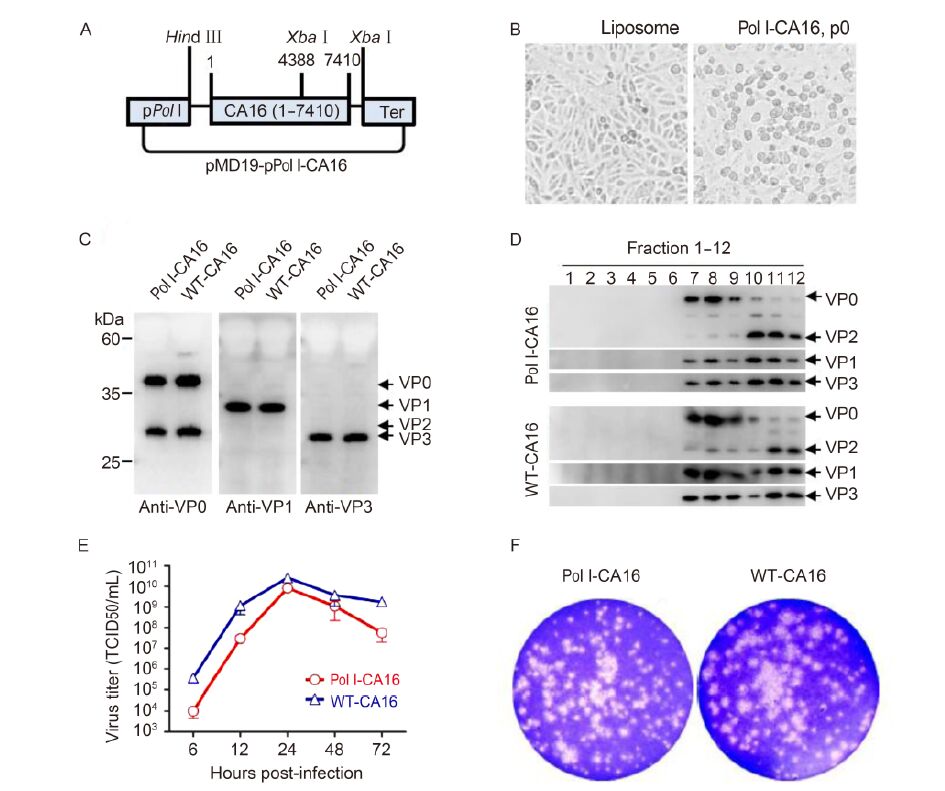
Figure 1. Generation and characterization of the recovered Pol Ⅰ-CA16 particles. (A) Construction of pMD19T-pPol Ⅰ-CA16 plasmid. Full-length CA16 genome (1-7410 bp) was flanked by Pol Ⅰ promoter (pPol Ⅰ) and murine terminator (Ter) sequence, between Hind Ⅲ and Xba Ⅰ restriction sites. (B) Cytopathic effect (CPE). Plasmid pMD19T-pPol Ⅰ-CA16 was used to transfect Vero cells. CPE was observed after 48 h, while the control cells grew normally. (C) Capsid composition. Lysates from cells infected with Pol Ⅰ-CA16, p6 or WT-CA16 were probed using CA16-specific antibodies (Liu et al., 2011). (D) Particle assembly. Pol Ⅰ-CA16, p6-or WT-CA16-infected cells were layered onto 10%-50% sucrose gradients and subjected to centrifugation (himac cp80wx, 111500g, 3 h at 4 ℃). Fractions (12, top to bottom) were collected and probed with antibodies by western blotting. (E) One-step growth curves. Vero cells were infected with Pol Ⅰ-CA16, p6 or WT-CA16 (MOI = 1.0). Infected cells were harvested 6, 12, 24, 48, and 72 h post-infection and TCID50 were determined on Vero cells. Statistical analyses were performed by the Student two tailed test using GraphPad Prism version 5. (F) Plaque phenotype. Plaque assay of Pol Ⅰ-CA16, p6 or WT-CA16 was done as described previously (Liu et al., 2011) and the results are shown.
A mixture of pMD19T-pPol Ⅰ-CA16(4 μg) and Lipofectamine 2000 reagent (Invitrogen) was used to transfect Vero (ATCC# CRL-1586) cells (Meng et al., 2012). Cytopathic effect (CPE) was observed 48 h post-transfection. Control cells appeared to grow normally, whereas plasmid-transfected cells displayed typical CPE (Figure 1B). The virus was harvested by repetitive freeze-thawing 72 h post-transfection. The infectious virus, named "Pol Ⅰ-CA16, p0", was efficiently recovered with high viral titer (Supplementary Figure S1A).
The recovered virus (Pol Ⅰ-CA16, p0) was blind-passaged (six rounds) on Vero cells (MOI = 0.02) to test its stability. Viruses from each passage were named Pol Ⅰ-CA16, p1-p6 accordingly. Viral titer (TCID50) of each generation was tested using the Reed-Muench method (Liu et al., 2011). TCID50 of the recovered Pol Ⅰ-CA16, p0 was as high as 8.95 × 108/mL, and remained rela-tively stable during passaging (Supplementary Figure S1A). Viral RNA was extracted for each passage. RT-PCR was performed using CA16-specific primers (Liu et al., 2011). Expected ~900-bp bands of VP1 gene (Supplementary Figure S1B) were detected in all samples. Western blotting with CA16-specific anti-VP1 antibody indicated that VP1 was correctly cleaved during serial passaging (Supplementary Figure S1C). Typical CPE was observed in cells infected with Pol Ⅰ-CA16, p1-p6 and wild-type virus (Supplementary Figure S1D). The sequence of the capsid-encoding gene P1 of Pol Ⅰ-CA16, p6 virus was identical to the wild-type, as confirmed by sequencing (data not shown). Collectively, these results suggested that the recovered virus was stable during passaging.
Biological characteristics and pathogenicity of the final recovered virus and wild-type virus were compared. Capsid composition of Pol Ⅰ-CA16, p6 virus was an-alyzed and no difference in viral protein production or processing between this virus and WT-CA16 was detected (Figure 1C). Sucrose gradient assembly analysis of the recovered virus suggested that it assembled in a particle form, as the wild-type (Figure 1D). The one-step growth curve was also analyzed. Vero cells were infected with the recovered or wild type virus (MOI = 1.0) (Meng et al., 2012). Infected cells were harvested at different time post-infection and TCID50 was determined on Vero cells. The wild-type virus and Pol Ⅰ-CA16, p6 virus have similar growth kinetics (Figure 1E). The plaque assay was performed as previously described (Liu et al., 2011), and plaque sizes of Pol Ⅰ-CA16, p6 and wild-type virus were similar (Figure 1F). These results suggested that the characteristics and pathogenicity of the recovered virus were similar to the wild-type virus.
Pol Ⅰ enzyme is the major form of nuclear DNA-dependent RNA polymerase in eukaryotic cells. It synthe-sizes ribosomal RNA precursors in the nucleolus (Hoffmann and Webster, 2000; Russell and Zomerdijk, 2006). Viral RNA and protein synthesis was investigated in pMD19T-pPol Ⅰ-CA16 plasmid-transfected Vero cells. Presumably, after entry into the cell, two types of RNA, differing in their non-coding regions, were synthesized from the plasmid template. Positive-sense RNA may be transcribed by cellular Pol Ⅰ, presumably in cellular nu-cleoplasm. A second positive-sense RNA type would be synthesized by viral polymerase after viral replication and transcription. Although both RNA types differ in 5′and 3′non-coding regions, they contain the same open reading frames encoding all viral proteins. Interaction of all nucleotides and proteins produced by cellular and vi-ral transcription and translation machineries then results in generation of infectious CA16 virus particles. Further study is required to elucidate details of viral replication.
The novel Pol Ⅰ promoter-driven reverse genetics system has the following advantages:(1) it is very simple: any laboratory with cDNA, plasmid, and transfection technologies can employ it; (2) RNA Pol Ⅰ is abundant in growing cells, ensuring high efficiency of viral DNA transcription, e.g., the system was successfully applied in infectious influenza virus research (Hoffmann et al., 2000); (3) compared with the T7 polymerase system, transcription of cDNA into RNA is more precise in the eukaryotic system (Neumann and Kawaoka, 2001; Sousa and Mukherjee, 2003).
In summary, we generated an improved plasmid-based recovery system for the CA16 infectious clone, driven by Pol Ⅰ promoter. Biological characteristics and pathogen-icity of the recovered virus are similar to wild-type CA16. Capsid assembly (Figure 1C, 1D), viral replication curve (Figure 1E), and phenotypic characteristics (Figure 1F) were also analyzed. The results suggested that the virus recovered after serial passaging was identical to the wild-type virus, indicating its stability and infectivity. This method constitutes a valuable tool for CA16 research.
-
We thank Chenghao Jin (University of Western Australia) for his editorial contribution in manuscript preparation. We thank Drs. Bing Sun and Qi Jin for providing the CA16 virus. This work was supported by grants from the Science and Technology Commission of Shanghai Municipality (13ZR1462900) and the Shanghai Institutes for Biological Science (SIBS), Chinese Academy of Science (CAS) (2013KIP317). Q. L. also acknowledges the support of the SA-SIBS scholarship program and Youth Innovation Promotion Association of CAS (2016249). This article does not contain any studies with human or animal subjects performed by any of the authors. The authors declare no conflicts of interest.
Supplementary figure/table are available on the websites of Virol-ogica Sinica: www.virosin.org; link.springer.com/journal/12250.
-

Figure S1. Stability of the recovered Pol Ⅰ-CA16 particles. (A) Viral titers of Pol Ⅰ-CA16. Each generation was titrated by TCID50 assay on Vero cells. Statistical analyses were performed with GraphPad Prism version 5. (B) Detection of Pol Ⅰ-CA16 negative-sense RNA. RNA was extracted from viral particles from each generation and negative-sense RNA was detected by RT-PCR. Lane M, DNA marker; lane p1-p6, PCR products from the recovered infective virus. (C) Viral capsid proteins of Pol Ⅰ-CA16. Proteins were detected using CA16 specific anti-VP1 antibody by western blotting (Liu et al., 2011). Lane p1-p6, Pol Ⅰ-CA16 recovered from infected cell lysates. (D) Recovered Pol Ⅰ-CA16 caused CPE upon passaging. The recovered Pol Ⅰ-CA16 virus or WT-CA16 (equivalent MOIs) was passaged on Vero cells over six generations, and each generation resulted in CPE, as presented.
Primer Sequence (5'-3') Enzyme site P1 ACCGCCGGGAGGGCGTCCCC None P2 

P3 GGCCGCAGATCTCCCCCCCAACTTCGGAGGTCGACCAGTACTCC Xba Ⅰ P4 GGCCGGAGTACTGGTCGACCTCCGAAGTTGGGGGGGAGATCTGC Xba Ⅰ P5 GCCAAGCTTAAAACAGCCTGTGGGTTGTTCCCACCC Hind Ⅲ P6 CGGGTCTAGAGCGTAGACTCTTTTGGCTTCAGTC Xba Ⅰ P7 CTGGTCTAGAAAGAAGGATGAACAACTAC Xba Ⅰ P8 TATTCTAGATTTTTTTTTTTTTTTTTTTTTTTTT Xba Ⅰ Table S1. Primers used in this study

 DownLoad:
DownLoad: