











-
The etiology and infectious pathology of herpes simplex virus 1 (HSV-1) has been studied in depth because it might infect close to 70% of the population at some point in each individual’s lifetime (Looker et al., 2015). The virus causes various symptoms, including herpes labialis, herpangina, herpetic encephalitis and herpes progenitalis, with increasing morbidity in young populations (Xu et al., 2006; Fatahzadeh and Schwartz, 2007; Garland and Steben, 2014). Data obtained from various sources indicate that its complicated genomic structure and the various biological features of its encoded proteins are involved in the viral pathogenesis observed in infected hosts (Kukhanova et al., 2014). Thus, by modifying specific viral genes, we can obtain mutants with different biological phenotypes that could serve as research models for a functional study of the corresponding genes. Studies analyzing the phenotypes of mutated HSV-1 strains with modifications to the ICP27, ICP0, gD, gK and many other genes support this correlation (Brehm et al., 1999; Awasthi et al., 2008; Strain and Rice, 2011; David et al., 2012) and provide data that could aid our understanding of the biological roles of viral-encoded proteins during infection.
Research focused on the molecules encoded by HSV-1 has indicated that tegument proteins associated with the viral structure and functional characteristics usually play specific roles in the virus–host interaction (Kelly et al., 2009; Xu et al., 2016a). More than 20 tegument proteins have been identified, but they have not been completely investigated. However, several studies have focused on the tegument protein VP16 and virion host shut-off (vhs; also known as UL41) protein due to their key roles in viral proliferation and infection. The results suggest that tegument proteins are important elements of HSV-1 and can determine specific viral phenotypes, and that they could be used to investigate viral pathogenesis during infection (Herrera and Triezenberg, 2004; Cotter et al., 2010; Saffran et al., 2010; Sawtell et al., 2011).
There are currently many promising vaccine approaches being explored to screen for a suitable vaccine type. Although many vaccine approaches have been investigated in animals and humans, live attenuated vaccines have distinct advantages over other vaccine types because they allow the expression of the entire repertoire of viral antigens (Stanfield and Kousoulas, 2015; Johnston et al., 2016). Accordingly, in this study, we produced attenuated viruses that might be candidates for vaccine development using CRISPR/Cas9 gene modification. In our previous study, we constructed an attenuated HSV-1 strain by partially deleting the UL7 gene, which encodes a tegument protein. The protein has been identified as constituting approximately 1% of the total HSV-1 tegument proteins (Tanaka et al., 2008; Loret and Lippé, 2012), and it might be involved in transcription/regulation of the viral α-gene and a factor restricting viral proliferation (Xu et al., 2016b). As expected, the partial deletion mutation of the UL7 gene leads to a viral strain that exhibits not only a lower proliferative rate but also an attenuated phenotype during infection in animals.
Thus, to acquire further attenuated strains that are more suitable for vaccine development, in this study, we chose the UL41 tegument protein as a second target for the design of an attenuated HSV-1 vaccine. The UL41 protein is capable of impairing activation of the RNA-sensing pathway involving Toll-like receptor 3 (TLR3) and RIG-I-like receptors (RLRs) in conventional dendritic cells (DCs) and preventing antigen signal transfer from the innate to the adaptive immune system during infection (Samady et al., 2003; Paludan et al., 2011). Furthermore, to restrain latent infection, the LAT gene, which is related to latency establishment and maintenance, was selected as the third gene to be modified.
Hence, in this study, UL7, UL41 and LAT mutations were each constructed using the CRISPR/Cas 9 method and then comparatively analyzed. Observations of a mutant stain with modified UL7 and UL41 genes (M2) indicated that it had a weaker virulence in animals compared with the strain with only a modified UL7 gene (M1). However, an analysis of latent viral infection involving tracing LAT expression in the trigeminal ganglion of infected animals using qRT-PCR led to positive results for both M1 and M2. Further deletion of a 138-nt fragment from LAT in the M2 mutant (to yield M3) resulted in a lower proliferative ratio in various cells, non-lethal infection in mice, a lower viral load in nervous tissues compared with the wild-type (WT) strain, and no LAT in the trigeminal ganglion of infected animals.
The results involving the three above-mentioned HSV-1 mutants suggest a possible approach for constructing a live attenuated mutant virus for use as an HSV-1 vaccine. In addition, comparative analysis of the three strains demonstrated that M3 exhibits the lowest pathogenicity during acute and latent infection, indicating that this mutant is an ideal candidate for HSV-1 vaccine development.
-
The human embryonic kidney (HEK) cell line 293T, the human glioma cell line Hs683, the human neuroblastoma cell line SH-SY5Y, the African green monkey kidney cell line Vero (ATCC, Rockefeller, Maryland, USA) and the human diploid cell line KMB17 (IMB, CAMS, Kunming, Yunnan, China) were maintained in Dulbecco’s modified Eagle medium (DMEM; HyClone, Logan, Utah, USA) with a high concentration of glucose supplemented with 10% fetal bovine serum (FBS) (DMEM; HyClone, Logan, Utah, USA). After viral infection, the culture medium was changed to DMEM supplemented with 2% FBS. The pathogenic WT HSV-1 strain 8F (Yu et al., 2010) and the mutant M1 strain (Xu et al., 2016b) were used in our experiments. The virus titers were determined using the 50% tissue culture infectious dose (TCID 50) method with Vero cells.
-
Two sets of guide RNAs (g-RNAs) were designed to target the UL41 and LAT genes according to the protocol reported by Ran et al. (2013) . The g-RNA sequences were named UL41-1-top, UL41-1-bottom, UL41-2-top, UL41-2-bottom, LAT-1-top, LAT-1-bottom, LAT-2-top and LAT-2-bottom. The genomic regions surrounding the CRISPR target sites of the genes were PCR-amplified with the primers UL41-F, UL41-R, LAT-F and LAT-R. The specific primer sets used are listed in Supplementary Table S1.
-
The g-RNA expression plasmids were constructed according to the protocol reported by Ran et al. (2013) and Xu et al. (2016b) . The g-RNA dsDNA fragments were annealed and inserted into the Bbs1 (New England BioLabs, Ipswich, MA, USA) site of the CRISPR/Cas9 system vector PX330 (Addgene, Cambridge, MA, USA). A pGM-T vector ligated to a UL30 gene fragment was used as the standard template for qRT-PCR. The plasmids were transfected into 293T cells using the Fugene HD reagent (Promega, Madison, WI, USA) according to the manufacturer’s recommended protocol. The specific primer sets used are listed in Supplementary Table S2.
-
For the construction of UL7-UL41-MU (M2), 293T cells were first co-transfected with the plasmids PX330-UL41-1 and PX330-UL41-2 and then infected with the M1 virus. The M2 virus, which has a partial UL41 deletion mutation, was harvested from the infected 293T cells at 48 hours post infection (h.p.i.), and viral genomic DNA was extracted using a TIANamp Virus RNA/DNA Kit (Tiangen, Beijing, China). The genomic region surrounding the CRISPR target site of the UL41 gene was PCR-amplified using PrimeSTAR DNA polymerase (TAKARA, Dalian, China) with the primers UL41-F and UL41-R. The products were analyzed on 1.5% agarose gels, which were stained with ethidium bromide (EB) and imaged using a Bio-Rad Gel Doc gel imaging system (Bio-Rad, California, USA). After detecting the mutation efficiency, the mutated virus was purified via a plaque assay.
For the construction of UL7-UL41-LAT-MU (M3), 293T cells were co-transfected with the plasmids PX330-LAT-1 and PX330-LAT-2 and then infected with the M2 virus. The M3 virus, which has a partial LAT deletion mutation, was harvested from the infected 293T cells at 48 h.p.i., and viral genomic DNA was extracted using the TIANamp Virus RNA/DNA Kit (Tiangen, Beijing, China). The genomic region surrounding the CRISPR target site of the LAT gene was PCR-amplified using PrimeSTAR HS DNA polymerase with GC buffer (TAKARA, Dalian, China) and the primers LAT-F and LAT-R. The products were analyzed on 1.5% agarose gels, which were stained with EB and imaged using the Bio-Rad Gel Doc gel imaging system. After detecting the mutation efficiency, the mutated virus was purified via a plaque assay.
-
For the plaque assay, 10-fold serial dilutions of the virus material ranging from 10–2 to 10–6 were prepared in DMEM without FBS. The HSV-1 WT, M1, M2 and M3 virus dilutions were grown in Vero cells overlaid with 0.6% agarose in growth medium at 37 °C. Four days post infection (d.p.i.), the cells were fixed and stained with 0.2% crystal violet in 20% ethanol, and the plaques were imaged to assess their morphology. KMB17, Hs683 and SH-SY5Y cells were infected with the HSV-1 WT, M1, M2 and M3 viruses at a multiplicity of infection (MOI) of 0.01 at 37 °C. The total virus molecules in the cell culture supernatants and the infected cells were harvested at several time points (12, 24, 36, 48, 60 and 72 h.p.i.), and the virus titers of all of the samples were determined using the standard TCID 50 virus titration method involving in Vero cells.
-
BALB/c mice were lightly anesthetized with ether and then infected by intranasal instillation of 2 × 10 5 plaque-forming units (PFUs) of the HSV-1 virus (WT, M1, M2 and M3) or sterile phosphate-buffered saline (PBS, pH 7.4) as a control (10 mice in each group). The weight of the mice was measured every 2 d, and the survival rate-to-mortality ratio was assessed over a 10-d period. Tissue samples from the mice were obtained immediately after death and used for assessments of organ pathology.
-
The mouse organs were fixed in 10% formalin and embedded in paraffin to obtain tissue blocks. Approximately two slides of each organ were stained with hematoxylin and eosin (H&E) for the assessment of morphology. For the immunohistochemical examinations, the specimens were immersed in 0.3% hydrogen peroxide in PBS for 10 min to block any intrinsic peroxidase activity. The specimens were blocked with 5% bovine serum albumin (BSA) in PBS at 37 °C for 15 min and then incubated with a rabbit polyclonal anti-HSV-1 antibody (Abcam, Cambridge, UK) at 4 °C for 12 h. After rinsing with PBS, the sections were incubated with a mixture of Peroxidase AffiniPure Rabbit Anti-Goat IgG (Abcam, Cambridge, UK) at 37 °C for 30 min. Enzyme immunohistochemistry was performed using the standard avidin-biotin-peroxidase method with 3, 3′-diaminobenzidine (DAB) as the substrate (Tiangen, Beijing, China) according to the manufacturer’s recommended protocol. After rinsing with water for 6 min, the sections were stained with hematoxylin for assessment of the cell nuclei. For each staining condition, a slide coated with rabbit IgG (Abcam, Cambridge, UK) was stained and examined in parallel as a control.
-
The viral load in the mice was determined by qPCR with absolute quantification. Based on the method developed by Kessler et al., the primers for the reaction were selected from a highly conserved region of the DNA polymerase gene UL30 in the HSV-1 genome, which would allow amplification of a 92-bp fragment of the HSV-1 DNA. A standard curve was simultaneously obtained using standard DNA samples (a pGM-T plasmid ligated to a UL30 gene fragment). Viral genomic DNA was extracted from the mouse tissues using a Universal DNA Purification Kit (Tiangen, Beijing, China). The TaqMan probe (Sangon Biotech, Shanghai, China) was labeled with 6FAM at the 5′ end and TAMRA at the 3′ end. The reactions were performed using Premix Ex Taq TM (Probe qPCR; Takara, Dalian, China) on an ABI 7500 thermocycler (Life Technologies, Carlsbad, CA, USA).
For the relative quantification of RS1 and LAT expression in the mice, the expression levels were calculated using the comparative Ct method (ΔΔCt) with the mouse housekeeping gene GAPDH. Gene expression was expressed as the fold change (2-ΔΔCt) relative to the levels in samples from the PBS-injected mice, which were used for calibration. The total RNA from mouse tissues was extracted using TRIzol reagent (Life Technologies, Carlsbad, CA, USA), and amplification reactions were performed using a One-Step SYBR Prime ScriptTM PLUS RT-PCR Kit (Takara, Dalian, China).
Template-negative and RT-negative reactions served as controls, and the specific primer sets used are listed in Supplementary Table S2.
-
An in situ hybridization assay was performed using an Enhanced Sensitive ISH Detection Kit I (POD) (Boster, Wuhan, China) according to the manufacturer’s recommendations. Briefly, the frozen sections were fixed with 4% paraformaldehyde for 20 min at room temperature. After rinsing with distilled water, the sections were immersed in 0.6% hydrogen peroxide in methanol for 30 min to block any intrinsic peroxidase activity. After three rinses with distilled water, the sections were subjected to pepsin digestion for 5 s and then treated with the pre-hybrid solution at 37 °C for 3–4 h.
Subsequently, 20 μL LAT probe (1 μg/mL in hybrid solution) was pipetted onto each sample, and the samples were incubated at 37 °C overnight in a humidity chamber. The sections were treated with sealing liquid for 30 min at 37 °C, mouse anti-DIG for 60 min at 37 °C, SABC solution for 20 min at 37 °C, anti-mouse-HRP-polymer for 20 min at 37 °C, and diaminobenzidine chromogen (DAB) for 10 min at room temperature. After rinsing with water for 6 min, the sections were stained with hematoxylin for assessment of the cell nuclei. LAT positivity was evaluated in terms of the whole cell, with any brown dot-like signal from a cell being considered indicative of LAT expression. Cells without a signal were considered negative for LAT expression.
-
The data from the various assays, which were performed in triplicate, are expressed as the mean values with standard deviations. GraphPad Prism software (San Diego, CA, USA) was used for the statistical analyses. The differences between each pair of groups were evaluated by one-way analysis of variance (ANOVA). A survival analysis was performed to analyze the survival rate of the infected mice.
-
Our previous study of an HSV-1 mutant with a partially deleted UL7 gene (M1) showed that there was a reduction in the dynamic growth associated with the mutated tegument protein, UL7 (Xu et al., 2016b). To develop a potential attenuated HSV-1 vaccine candidate, the M1 strain was used as the basic strain for further mutations of the UL41 and LAT genes. This involved a 30-bp (225–254) sequence of the UL7 gene, a 59-bp (375–433) sequence of the UL41 gene, and a 138-bp (937–1074) sequence of the LAT gene, which were deleted sequentially using the CRISPR/Cas9 method. The mutants were identified by PCR followed by sequencing of the PCR products, and the mutated clones were acquired via plaque screening in Vero cells (Figure 1).

Figure 1. Design of the HSV-1 M1, M2 and M3 strains and verification of their mutant genes. (A) Flowchart of the design and construction of the M1, M2 and M3 viruses. (B) Identification of the mutated UL7 gene in the purified M1 virus. The UL7 mutation (225–254, 30-bp deletion) was identified via PCR using UL7-sense and UL7-antisense primers. (C) Identification of the mutatedUL41 gene in the purified M2 virus. The UL41 mutation (375–433, 59-bp deletion) was identified via PCR using the UL41-F and UL41-R primers. (D) Identification of the mutatedLAT gene (937–1074, 138-bp deletion) in the purified M3 virus. The LAT mutation was identified via PCR using the LAT-F and LAT-R primers.
The HSV-1 genome is flanked by a pair of inverted repeat regions (terminal repeat long [TRL]/internal repeat long [IRL] and internal repeat short [IRS]/terminal repeat short [TRS]), and there are two copies of theICP34.5, ICP0 and LAT genes, in the TRL regions. Therefore, we used PCR and sequencing to confirm that both copies of the LAT gene in different generations of M3 viruses (P1–P15) were successfully knocked out. Subsequently, we completed the preliminary whole-genome sequencing of the M3 strain to further determine the gene mutations, and we identified mutations in three genes (UL7, UL41 and LAT) (Supplementary Figure S1–S3). In addition, no off-target mutations were observed based on sequence alignment. Finally, the M2 strain (with mutated UL7 and UL41 genes) and the M3 strain (with mutated UL7, UL41 and LAT genes) were produced to carry out a comparative study of the two strains together with the M1 strain.
-
The three mutant strains obtained via plaque selection were first analyzed based on their plaque characteristics in Vero cells, which revealed a downward trend in the viral growth rate from WT to M1 and from M1 to M2 and M3. Under identical experimental conditions, including the same culture temperature and viral MOI, the three mutant strains exhibited similar characteristic plaque shapes, which were different from that of the WT strain (Figure 2A). The plaques induced by the WT strain were notably larger than those obtained with the three mutant strains, and the plaques induced by the M2 and M3 strains were smaller than those produced by the M1 strain (Figure 2A). This result indicated that the mutation of the UL41 gene enhanced the effect of the mutated UL7 gene, and that the mutation of the LAT gene might not influence viral proliferation in non-neuronal cells, suggesting that only the UL7 and UL41 genes are involved in the regulation of viral replication.
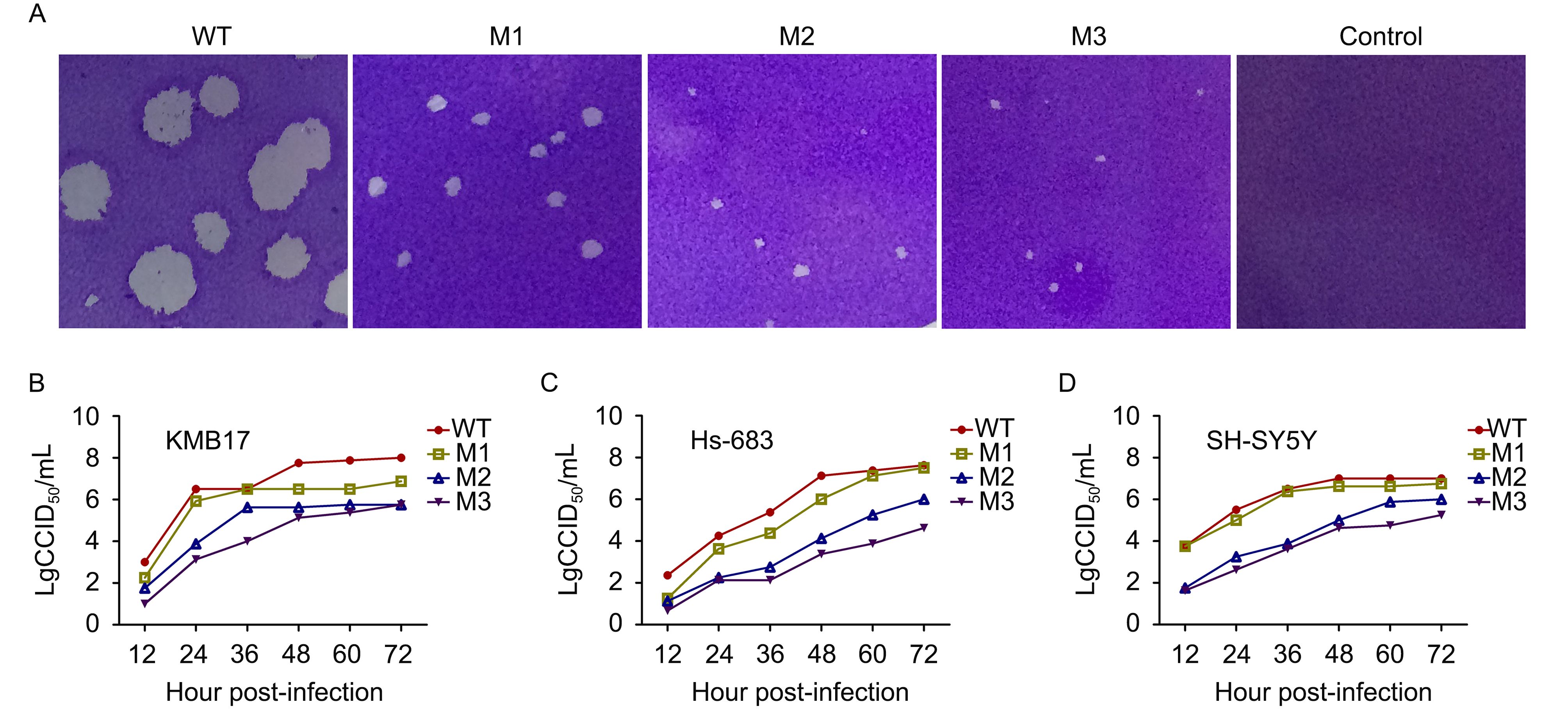
Figure 2. Plaque phenotypes and dynamic growth characteristics of the HSV-1 WT, M1, M2 and M3 strains in cultured cells. (A) Plaque phenotypes of the WT, M1, M2 and M3 strains in Vero cells. Vero cells were infected with the four viruses or DMEM as a control at an MOI of 0.0001 and were then fixed and stained at 4 d.p.i. Dynamic profiles of the four strains in (B) KMB17, (C) Hs683 and (D) SH-SY5Y cells. KMB17, Hs683 and SH-SY5Y cells were infected with the WT (filled circles), M1 (open squares), M2 (upward open triangles) or M3 (downward filled triangles) strains at an MOI of 0.01. The supernatants and cells were harvested at the indicated time points, and the lysates were titrated in Vero cells. The error bars indicate the standard deviations (SDs). The data are shown as the means ± SDs based on three independent experiments.
Further observation of the dynamic growth of these strains in the human fibroblast cell line KMB17 (Figure 2B) indicated that all three mutant strains, particularly M2 and M3, possess a lower proliferative capacity than the WT strain. Moreover, the dynamic growth of the mutant strains in the human glioblastoma cell line HS683 (Figure 2C) and the human neuroblastoma cell line SH-SY5Y (Figure 2D) revealed that the M2 and M3 strains had a low proliferation capacity. For example, in HS-683 cells, the M3 titer reached lg4.0 CCID 50/mL at 72 h.p.i., whereas the WT titer reached lg7.0 CCID 50/mL at the same time point (Figure 2C). These data further suggested that the modifications to the UL7 and UL41 genes altered the viral proliferative efficacy, and that M2 and M3 could be characterized as strains with slow proliferation phenotypes and they could be further investigated as such.
-
Based on the lower proliferative capacity of the three mutant strains described above, an animal experiment was performed to investigate the possible correlation of this lower proliferation with virulence in infected mice. Fifty BALB/c mice were divided into five groups, and the mice in each group were inoculated via nasal instillation with one of the following: the WT strain, one of the three mutants, or PBS. Daily records of post-infection clinical manifestations revealed that the weight of the mice infected with the WT strain decreased gradually, whereas the weight of the mutant- and PBS-infected mice increased gradually (Figure 3A). The mice infected with the WT strain began dying from 5 d.p.i., and 80% died by 8 d.p.i. In contrast, the survival rate of the mice inoculated with one of the mutant strains or PBS was 100% (Figure 3B), and no difference was observed between the M1, M2 and M3 strains.
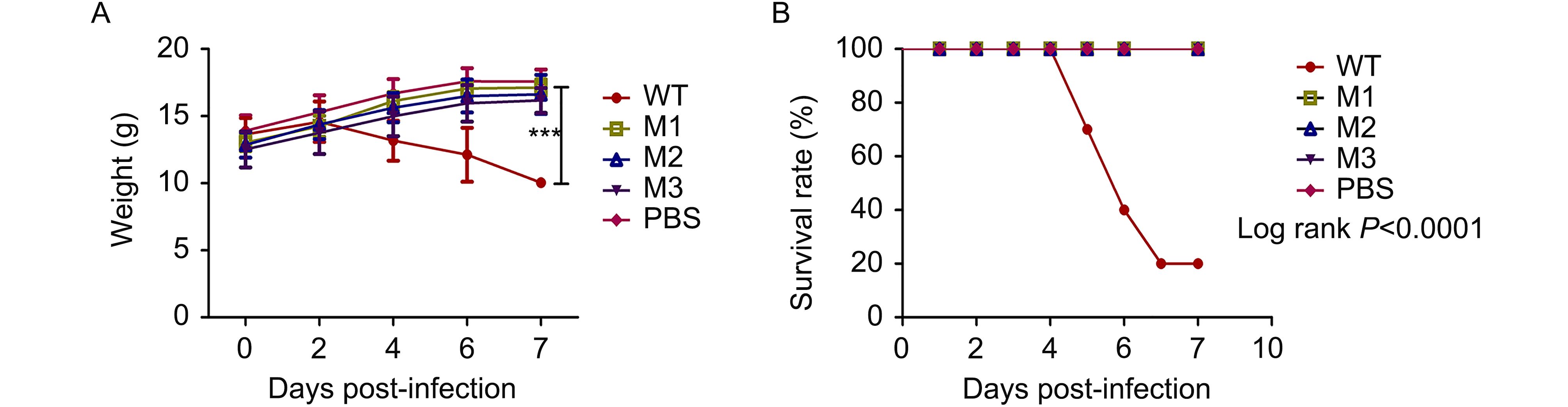
Figure 3. Attenuated phenotype of the HSV-1 M1, M2 and M3 strains in mice. (A) BALB/c mice were infected by intranasal instillation of 2 × 10 5 PFUs of the WT (filled circles), M1 (open squares), M2 (upward open triangles) or M3 (downward filled triangles) strains or PBS (filled diamonds) as a control (n = 10 for each group). The survival rates of the mice infected with the viruses are shown. (B) The weights of the mice were measured every 2 d. The data are shown as the means ± SDs. ***P < 0.005.
-
Previous data from histopathologic observations of mice infected with HSV-1 indicate that the infected animals usually die as a result of lesions in tissues of the central nervous system (CNS) (Dumitrascu et al., 2014). In the current study, histopathologic observations of mice infected with the WT strain indicated similar results. The observed encephalitis lesions involved the infiltration of inflammatory cells in several areas of the brain, particularly near vascular areas, accompanied by degeneration and necrosis of some of the neurons associated with hyperemia and even damage to the meninx structure (Figure 4A). Conversely, with the exception of a small degree of infiltration of inflammatory cells in some areas of the brain (including near vessels) (Figure 4A), no obvious neuronal degeneration or necrosis or other tissue damage was detected in the mice infected with the M1, M2 or M3 strains.
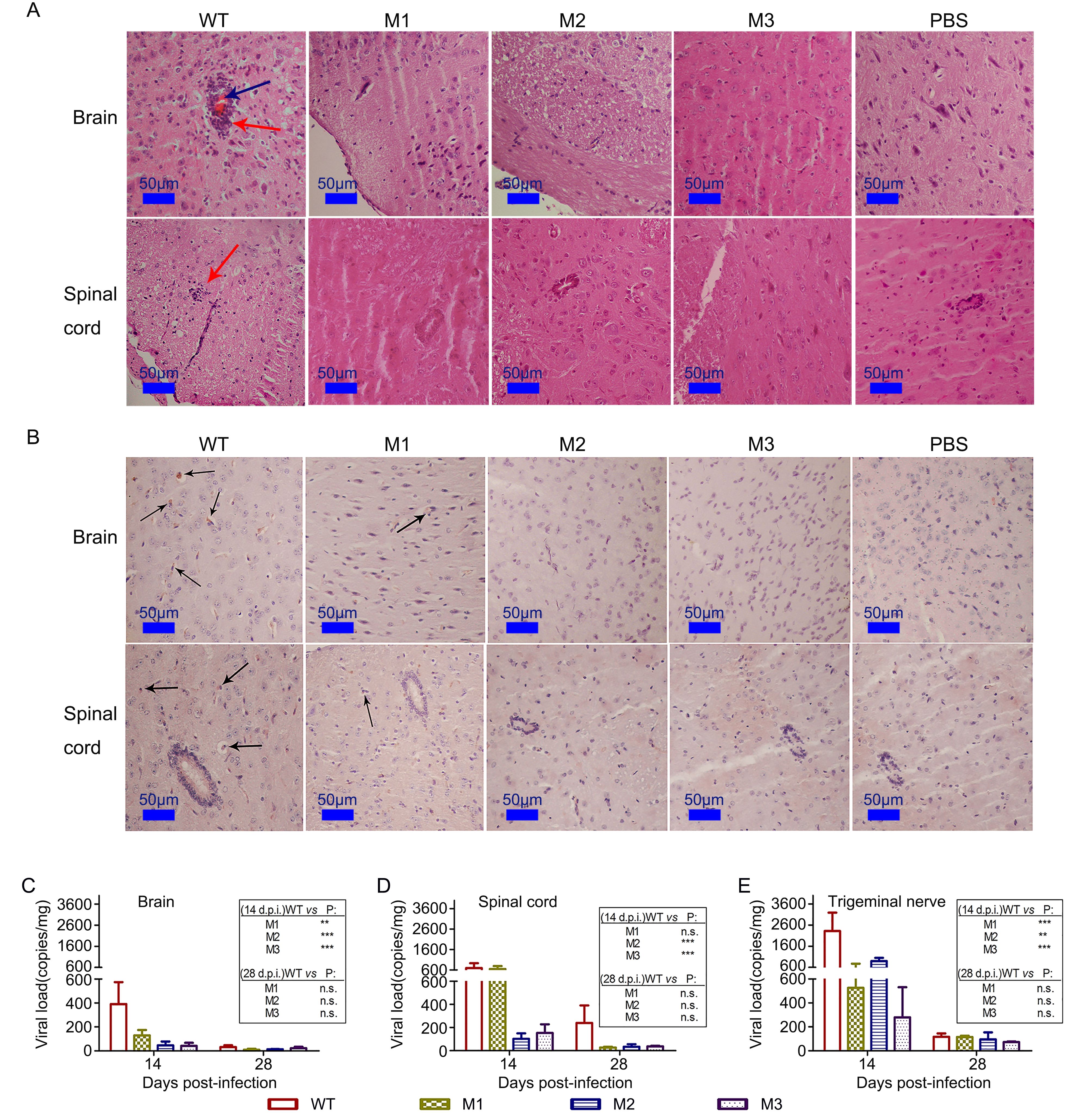
Figure 4. Histopathologic examination of CNS tissue of mice infected with the mutant and WT strains. (A) Pathological changes in the brain and spinal cords of mice infected with the WT, M1, M2 or M3 strains or PBS. The tissue sections were stained with H&E and imaged using an optical microscope at × 200 magnification. Tissue hyperemia is highlighted with blue arrows, and infiltration of inflammatory cells is highlighted with red arrows. (B) Immunohistochemical detection of HSV-1 in the brain and spinal cords from WT-, M1-, M2-, M3- or PBS-infected mice. Non-specific staining was observed in the PBS-infected mice. Positive expression of the HSV-1 antigen was detected in the brain and spinal cord tissues (arrows) of WT-, M1-, M2- and M3-infected mice. Assessment of viral load in the (C) brain, (D) spinal cord or (E) trigeminal nerve of mice challenged with WT, M1, M2 or M3, as determined by RT-qPCR. The viral copy numbers were quantified according to an HSV-1 DNA standard. The data are shown as the means ± SDs based on data from three independent mice.**P < 0.01; ***P < 0.005.
In the CNS tissue of the mutant-infected mice, an immunohistochemistry assay involving an antibody against HSV-1 showed slight viral antigen expression in areas infiltrated by inflammatory cells (Figure 4B), whereas obvious viral antigen expression was observed in the CNS of the mice infected with the WT strain (Figure 4B). In parallel, quantitative detection of the viral genomic DNA in these tissue samples (using qPCR) revealed that the viral loads in the WT-infected mice were 5- to 10-fold higher than that in the mutant-infected mice at 14 d.p.i. (Figure 4C–4E).
-
Previous data on HSV-1 infection in mice indicate that acute HSV-1 infection usually leads to pathological latent viral infection of nervous tissue (Aranda and Epstein, 2015), particularly the trigeminal nerve, as indicated by the presence of transcripts of the viral LAT gene (Wagner et al., 1988; Farrell et al., 1991). In the current study, we assessed the viral genomic DNA levels (using qPCR to detect the levels of the RS1 gene) in nervous tissues from mice infected with one of the three mutants or the WT strain. The results suggested that the trigeminal nerves of the mice infected with one of the mutant strains had lower viral loads than those of the mice infected with the WT strain (Figure 5A).
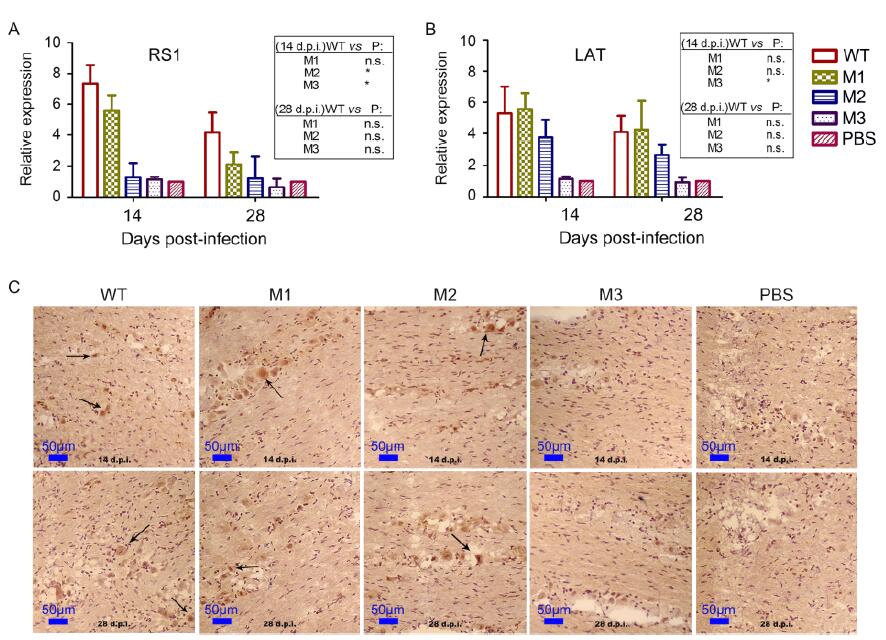
Figure 5. The three mutants showed a lower capacity for latent infection of the nervous system of mice compared with the WT strain. Levels of (A) RS1 and (B) LAT expression in the trigeminal nerve of mice challenged with WT, M1, M2, M3 or PBS at 14 or 28 d.p.i., as determined by relative RT-PCR. The graphic indicates the fold change in the RNA levels detected in virus-infected mice compared with PBS-injected mice. The RNA levels of the housekeeping gene GAPDH were used to normalize the target RNA levels detected in the mouse tissues. Relative quantification was performed using the comparative Ct method (△△Ct), with the RNA from the PBS-infected mice being used for calibration. (C) Positive LAT RNA expression, as determined using chromogenic in situ hybridization (× 200 magnification), is highlighted with black arrows. Brown-dot positivity can be observed for the WT-, M1- and M2-infected mice, with no positive signal for the M3- or PBS-infected mice.
Importantly, further tracing of LAT in the trigeminal ganglion of mice (based on qRT-PCR using specific primers with GGTCGGACGGGTAAGTAACAGAGT and CGCCCCAGAGGCTAAGGT sequences) revealed the presence of lower viral mRNA loads in mice infected with the M1 or M2 strains compared with the mice infected with the WT strain (Figure 5B), while the viral mRNA loads detected in the mice infected with the M3 strain were lower than the threshold of detection (Figure 5B). To confirm this finding, in situ hybridization with a specific probe was used, and it yielded a similar result (Figure 5C). These results suggested that the M3 strain might act as an attenuated strain that can restrict the establishment of latent infection in the trigeminal ganglion in mice.
-
In previous studies of HSV-1, viral mutants were constructed to investigate the biological roles of viral proteins and to select candidates for a viral vaccine (Morrison and Knipe, 1997; Koelle and Corey, 2003; Augustinova et al., 2004; Awasthi et al., 2008). In 2008, Takana et al. constructed an HSV-1 mutant with a deletion of the UL7 gene using the bacterial artificial chromosome (BAC) method, and they observed that the mutant had a lower proliferative capacity. The data obtained by Read et al. (1993) also show a similar low replication rate for a strain with a mutation in the UL41 gene. Based on these data, our previous work investigated the role of UL7 during viral proliferation and suggested that this molecule has a role in viral RNA transcription, as mutation of the UL7 gene led to an attenuated viral phenotype in an animal model of infection (Xu et al., 2016b). This previous research provided an initial carrier strain for further construction of attenuated vaccine candidates. Accordingly, the current study was designed based on an understanding of the biological functions of UL7, UL41 and LAT during viral infection.
It was presumed that specific modification focusing on the UL7, UL41 and LAT genes might create a strain with lower virulence, less interference ability to stimulate the innate immune system and reduced latent infection capacity in vivo. Thus, in this study, a preliminary analysis of the M1 mutant and two other mutants based on M1 (produced using the CRISPR/Cas9 method) was performed, focusing on their biological characteristics and pathogenicity in vivo. As indicated by the results, both the M1 and M2 mutants had a lower proliferative capacity and lower virulence in mice. The attenuated phenotype of the mutants provided us with a possible approach for further vaccine research. However, the mutated tegument proteins UL7 and UL41 induced little change in viral latency, suggesting the need to develop a new mutant with an attenuated phenotype with respect to both acute and latent infections, which required modification of the LAT gene. Previous data indicate that the LAT gene, which is highly expressed during viral latency, can significantly alter the degree of latent infection establishment in mice if mutated (Leib et al., 1989; Maggioncalda et al., 1994; Thompson and Sawtell, 1997; Samoto et al., 2001; BenMohamed et al., 2015). We found that mutation of the LAT gene combined with modified tegument genes (UL7 and UL41) can change the viral phenotype observed during animal infection, leading to very minor pathological lesions in the host nervous tissue and greatly decreased viral latency in the trigeminal ganglion.
An ideal live attenuated HSV vaccine candidate should possess characteristics that can generate robust immune responses while minimizing potential virulence in the hosts. In general, modification of genes that play important roles in the virus lifecycle may lead to lower pathogenicity and more robust production of humoral and cellular immune responses. Several attenuated strains with multiple deletions in the ICP0, ICP4, ICP22 and ICP47 genes or the UL29 and UL41 genes have been reported to induce immune responses and protect mice from WT HSV-1 challenge (Brehm et al., 1999; Geiss et al., 2000). However, these vaccine approaches face two major obstacles before they can be successfully used in humans: 1) Replication-defective viruses typically need to be grown in a complementing cell line, which may carry a cancer risk. The preparation method directly limits the widespread use of such strains. 2) Viruses with specific gene deletions are capable of entering into neurons and may establish latent infections, or potentially recombine with endogenous HSVs (Stanfield and Kousoulas, 2015). The mutant M3 strain obtained in the current study exhibited an attenuated phenotype, including lower virulence during acute infection, a very low degree of latent infection in mice and similar potential for use in vaccine development compared with other mutated HSV-1 strains. Furthermore, the mutant M3 strain was able to replicate and package complete viral particles in a variety of cell lines including Vero and diploid cells (KMB17). Although further understanding the properties of UL7 and the role played by the mutated UL7 in combination with reduced expression of UL41 or LAT will be important, the M3 strain provides a tool for future studies and for the development of an attenuated HSV-1 vaccine. Certainly, more work on the immunogenicity and safety of this strain will be needed.
-
This work was supported by the National Basic Research Program (2012CB518901), Chinese academy of medical sciences (CAMS) Initiative for Innovative Medicine (2016-I2M-1-019), the National Natural Science Foundation of China (31300143, 31100127), and the Fundamental Research Funds for the Central Universities (2016ZX310047, 2016ZX350072). The funders had no role in the study design, data collection and analysis, decision to publish, or preparation of the manuscript.
-
The authors declare that they have no conflict of interest. The experimental protocols were reviewed and approved by the Yunnan Provincial Experimental Animal Management Association (approval number: SCXK [Dian] 2011–0005) and the Experimental Animal Ethics Committee of the Institute of Medical Biology, Chinese Academy of Medical Sciences. The animal experiments were designed based on the principles expressed in the “Guide for the Care and Use of Laboratory Animals” and “Guidance for Experimental Animal Welfare and Ethical Treatment.” All institutional and national guidelines for the care and use of laboratory animals were followed.
-
QHL designed the experiments; XLX, STF, PFC, LCW and XLZ performed the experiments; XLX, YZ and YL performed the analyses; XLX and MF wrote the manuscript; YQG completed the majority of the manuscript revisions and refined the English used in the manuscript, and all of the authors contributed to, read, and approved the final version of the manuscript. Supplementary figures/tables are available on the websites of Virologica Sinica: www.virosin.org; link.springer.com/journal/12250.
-

Table S1. DNA sequences of g-RNAs and primers used for PCR

Table S2. Primers used for plasmid construction and qPCR

Figure S1. UL7 gene sequence alignment results

Figure S2. UL41 gene sequence alignment results
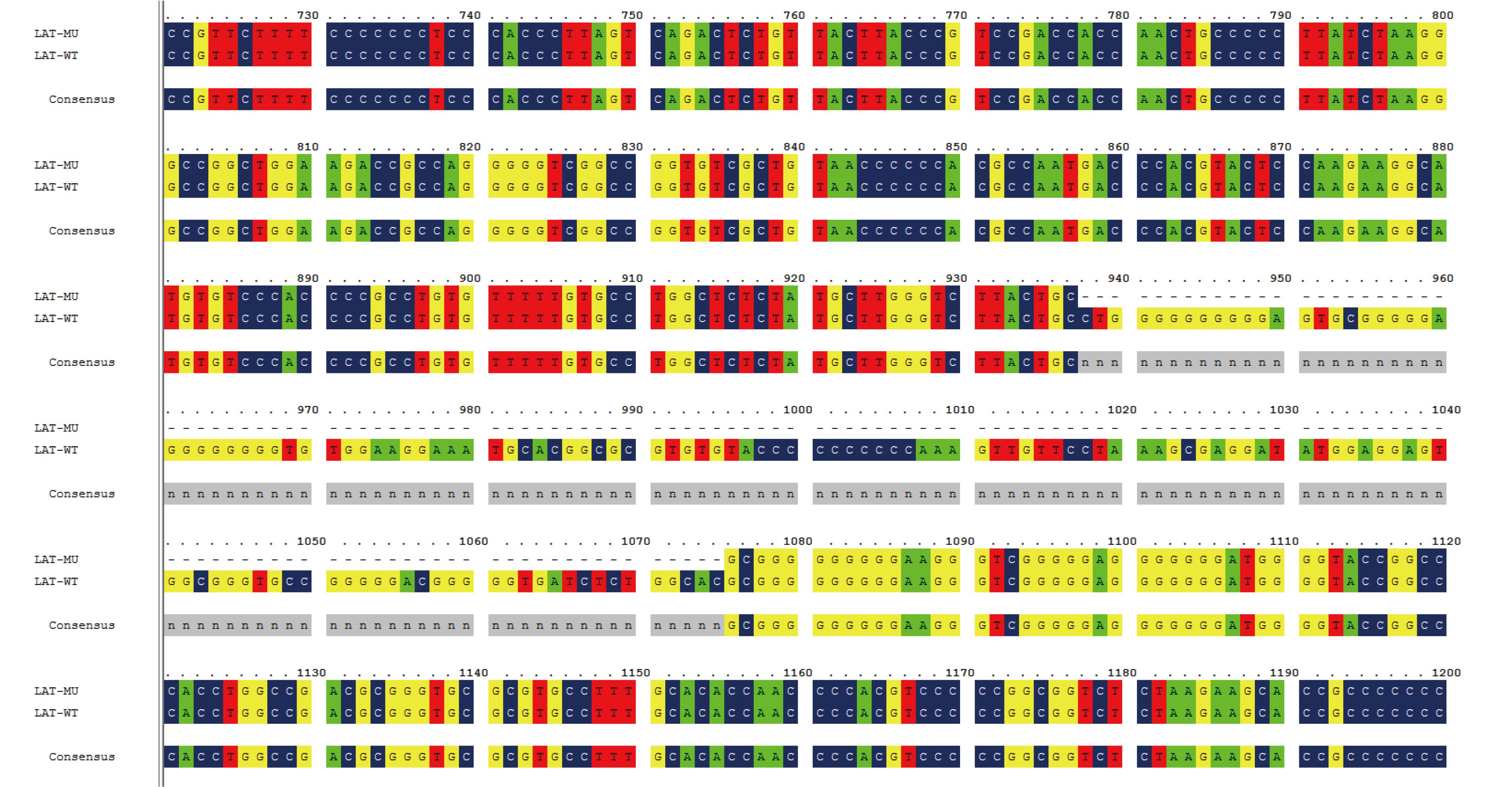
Figure S3. LAT gene sequence alignment results
Attenuated phenotypes and analysis of a herpes simplex virus 1 strain with partial deletion of the UL7, UL41 and LAT genes
- Xingli Xu # ,
- Yingqiu Guo # ,
- Shengtao Fan ,
- Pingfang Cui ,
- Min Feng ,
- Lichun Wang ,
- Ying Zhang ,
- Yun Liao ,
- Xiaolong Zhang ,
-
Qihan Li
,

- Received Date: 19 January 2017
- Accepted Date: 17 August 2017
- Published Date: 29 September 2017
Abstract: We previously constructed a herpes simplex virus 1 (HSV-1) UL7 mutant virus (M1) and showed that a partial deletion mutation of the UL7 gene led to a lower proliferative rate and an attenuated phenotype. Using the M1 mutant, we further modified the UL41 gene, which encodes another tegument protein, and the latency-associated transcript (LAT) gene. Observations of the resulting mutants with modified UL7 and UL41 (M2) or UL7, UL41 and LAT (M3) genes indicated attenuated phenotypes, with lower proliferative ratios in various cells, non-lethal infections in mice and lower viral loads in nervous tissues compared with the wild-type strain. Furthermore, no LAT stable intron could be detected in the trigeminal ganglion of M3-infected animals. The results obtained with the three HSV-1 mutants indicate that the M3 mutant is an attenuated strain with low pathogenicity during both acute and latent infections. Together, the results support the use of the M3 mutant as a candidate for the development of an HSV-1 vaccine.

 DownLoad:
DownLoad: