











-
Influenza A virus (IAV) is one of the most important pathogens to cause severe infectious disease among human and animals. IAV is a member of the Orthomyxoviridae family, and its genome is constituted by eight single-stranded negative-sense RNA segments. Due to the segmented nature of viral genome, IAV has the potential to mutate or reassort into new strains and spread quickly during seasonal epidemics (Everett et al., 2013; Zhu et al., 2013; Huang et al., 2017). Although our knowledge of the life cycle of IAV has increased tremendously, the molecular mechanisms that IAV uses to escape from innate immunity are not yet fully understood.
An increasing number of studies have shown that microRNAs (miRNAs) play important roles in antiviral response. miRNAs are non-coding, single-stranded, approximately 22-nucleotide RNAs that function as multifunctional regulators in animals and plants by targeting mRNAs for degradation or translational repression (Baltimore et al., 2008; O’Connell et al., 2010). Recent studies have identified several host-coded miRNAs that involved in IAV infection and replication through directly targeting the viral genome or regulation of type I interferon (IFN)-associated pathways (Song et al., 2010; Gui et al., 2015; Ingle et al., 2015). In turn, IAV infection could induce distinct cellular miRNA expression patterns. miR-26a has been identified as one of the significantly downregulated circulating miRNAs in the blood of influenza A H1N1 patients (Tambyah et al., 2013). But the molecular mechanism of the repression of miR-26a expression and the functional role of miR-26a upon IAV replication are not clear.
Innate immunity is the first line of defense against invading pathogens including IAV. IFNs are among the first molecules synthesized by IAV-infected cells. IAV infections trigger the type I IFN signaling pathway, leading to the transcription of hundreds of interferon-stimulated genes (ISGs) which exert antiviral effector functions (Zhou et al., 2011). Because this pathway limits replication and spread of IAVs, IAVs do not only induce type I IFN, but also antagonize the production and antiviral effects of IFN and ISGs. For example, IAV suppresses IFN-β production via activation of the viral non-structural protein1 (NS1) (Gack et al., 2009; Gao et al., 2012; Feng et al., 2017), but also inhibits type I IFN signaling through induction of the suppressor of cytokine signaling-3 (SOCS-3) protein (Pauli et al., 2008). Viral infection-induced miRNAs have been shown to regulate type I IFN signaling and subsequent anti-viral innate immunity (Zhou et al., 2011). Whether the dyregulated miRNAs by IAV infection are involved in the regulation of IFN signaling and the underlying mechanisms are not fully characterized.
In the present study, we found that overexpression of miR-26a in cells inhibits IAV replication through activating IFN signal pathway. However, IAV infection leads to markedly decreased expression of miR-26a. Thus, miR-26a is a protective factor against IAV replication. And downregulation of miR-26a may be a new strategy evolved by IAV to counteract cellular antiviral responses.
-
Human embryonic kidney cell line 293T, human alveolar epithelial cell line A549 and Madin-Darby canine kidney cell line MDCK were cultured in Dulbecco’s modified Eagle’s medium (DMEM) (Gibco, Paisley, UK) supplemented with 10% heat-inactivated fetal bovine serum (Gibco, Paisley, United Kingdom) at 37 °C in a humidified 5% CO 2 incubator.
Influenza virus A/WSN/33 (H1N1) was generated using 12-plasmid IAV reverse genetic system and propagated in MDCK cells (Neumann et al., 1999). Viral titer was measured using hemagglutinin (HA) assay or a standard plaque assay as described (Gao et al., 2015).
-
The mouse anti-influenza A NP (sc-101352), anti-USP3 (sc-135597) antibodies and NF-κB inhibitor Bay11-7082 were purchased from Santa Cruz Biotechnology (Santa Cruz, CA). The mouse anti-β-actin antibody was supplied by Sungene Biotech Co. (Tianjin, China). miR-26 mimic, mimic control, miR-26 inhibitor and inhibitor control were purchased from Guangzhou RiboBio Co., Ltd (Guangzhou, China).
-
The eight segments of IAV (HA, NA, NP, NS, PA, PB1, PB2, and M) were amplified from influenza virus A/WSN/33 using PCR and cloned into the 3′-UTR of the luciferase gene in the pRL-TK vector (Promega, Madison, WI) as described previously (Song et al., 2010). A 200-bp fragment of USP3 3′-UTR containing the predicated miR-26a binding site (position 62–68) and its mutant sequence were obtained by PCR and cloned into the Xba I site of pRL-TK vector. The plasmids encoding NF-κB p65 subunit, retinoic acid-inducible gene 1 (RIG-I), virus-induced signaling adapter (VISA), TANK-binding kinase 1 (TBK1), interferon regulatory factor 3 (IRF3) and IFN-stimulated response element (ISRE) reporter vector were provided by Pro. Xin Ye (Institute of Microbiology, Chinese Academy of Sciences, Beijing, China). The NF-κB-Luc reporter plasmid was a gift from Prof. Linbai Ye (College of Life Science, Wuhan University, Wuhan, China). The plasmid encoding Myc-USP3 was obtained from Sino Biological Inc (Beijing, China).
-
The plasmids and miRNA mimic/inhibitor were transfected into cells using Lipofectamine 2000 (Invitrogen, Carsland, CA) according to manufacturer’s instructions.
For IAV infection, A/WSN/33 virus was diluted in DMEM containing 2 μg/mL trypsin and added to the surface of 293T or A549 cells at the indicated multiplicity of infection (MOI). After 1-hour incubation, cells were washed with PBS to remove non-adherent virus and incubated for the duration of the experiment.
-
Total RNA was extracted with TRNzol reagent (TIANGEN Biotech, Beijing, China).
For miR-26a analysis, total RNA was reverse transcribed into cDNAs with miRNA first-strand cDNA kit (TIANGEN Biotech, Beijing, China). Quantitative real-time PCR was performed by using miRcute miRNA qRT-PCR detection kit (TIANGEN Biotech, Beijing, China) following manufacturer’s instruction. U6 was used as an internal control for normalization.
For viral RNA analysis, total RNA from A/WSN/33 virus-infected cells was used to perform reverse transcription using Revert Aid First Strand cDNA Synthesis kit (Thermo Scientific, Waltham, MA) with specific primers: 5′-AGCGAAAGCAGG-3′ and 5′-AGCAAAAGCAGG-3′. qRT-PCR was conducted using SYBR premix Ex Taq II (Takara Co., LTD, DaLian, China). 18S RNA was used as an internal control for normalization. The primer sequences used in the qRT-PCR were as follows: NA forward 5′-ATTCAAGGGGGACCTTTAAG-3′ and reverse 5′-CTGACCAAGCAACCGATTCA-3′; PB1 forward 5′-GCCACTGCCAGAAGACAA-3′ and reverse 5′-CGTTTCAAGACACGAGGT-3′; M1 forward 5′-ACAGAGACTTGAAGATGTCT-3′ and reverse 5′-CTAAAATCCCCTTAGTCAGA-3′; 18S rRNA forward 5′-CCATCCAATCGGTAGTAGCG-3′ and reverse 5′-CCCCAATGTCTCTGTTGTTGAC-3′.
For mRNA analysis, cDNA was synthesized from total RNA using Revert Aid First Strand cDNA Synthesis kit and qRT-PCR was performed by using SYBR premix Ex Taq II. GAPDH was used as an internal control for normalization. The primer sequences used in the qRT-PCR were as follows: IL-8 forward 5′-CTGCGCCAACACAGAAATTAT-3′ and reverse 5′-CATCTGGCAACCCTACAACAG-3′; IFN-α forward 5′-GTCAGAGTGGAAATCCTAAG-3′ and reverse 5′-ACAGCATCTGCTGGTTGAAG-3′; IFN-β forward 5′-GACTCCATCTTGGCTGTGA-3′ and reverse 5′-TGATTTCTGCTCTGACAACCT -3′; ISG15 forward 5′-TGGACA AATGCGACGAACC-3′ and reverse 5′-CCCGCTCACTTGCTGCTT-3′; GAPDH forward 5′-GGAGA AACCTGCCAAGTATG-3′ and reverse 5′-TTACTCCTTGGAGGCCATGT AG-3′.
PCR reactions were conducted in 7300 Real-Time PCR System (Applied Biosystems, Foster City, CA). The cycle conditions included an initial denaturation step at 95 °C for 2 minutes followed by 40 cycles of amplification for 15 seconds at 95 °C and 1 minute at 60 °C. Relative expression for mRNA was determined using 2 –ΔΔCt method. All reactions were performed in triplicate.
-
Total proteins were extracted from the transfected or virus-infected cells using cell lysis buffer (Cell Signaling Technology, Danvers, MA). The extracts were separated by SDS-PAGE and transferred to a nitrocellulose membrane (Amersham Biosciences, Amersham, UK). The membrane was blocked with 5% non-fat milk in Tris-buffered saline with Tween-20 (TBS-T) at room temperature for 2 h followed by incubation with the primary antibody at 4 °C overnight. After washing three times with TBS-T for 10 min each time, the membrane was incubated with a horseradish peroxidase-conjugated secondary antibody for 2 h at room temperature. Bands were detected using enhanced chemiluminescence (Applygen, China).
-
293T cells were transfected in a 24-well plate with miR-26a mimic (100 nmol/L) or inhibitor (100 nmol/L) together with 400 ng of the reporter constructs and 20 ng of pRL-TK control vector as indicated followed by infection with A/WSN/33 virus or left untreated. After incubation for the duration of the experiment, cells were collected and the luciferase activity was measured using Dual-luciferase Assay System (Promega, Madison, WI) according to manufacturer’s instructions. Each set of assays was performed in triplicate.
-
293T cells were transfected in a 12-well plate with miR-26a mimic (100 nmol/L) or its inhibitor (100 nmol/L) for 36 h followed by infection with A/WSN/33 virus at an MOI of 1 for 16 h. The concentration of IFN-β in the supernatants was measured by ELISA (PBL Assay Science, NJ, USA), according to the manufacturer’s protocol.
-
Data are presented as mean ± standard deviation (S.D.). Student’s t test was used to evaluate individual differences between groups. Analysis of variance (ANOVA) was used for multiple comparisons. P < 0.05 was considered to be statistically significant.
-
miR-26a has been reported to be significantly down-regulated in the blood samples of influenza patients (Tambyah et al., 2013). However, the molecular mechanism involved in miR-26 regulation and the role of miR-26a during IAV infection are not well understood. Here, decreased expression of miR-26a was also observed in IAV-infected 293T and A549 cells (Figure 1A). Furthermore, IAV (A/WSN/33) infection reduced miR-26a in a dose-dependent manner (Figure 1B). We then explored the molecular mechanism by which miR-26a was downregulated. It was reported that IAV infection activates NF-κB significantly (Flory et al., 2000). Moreover, NF-κB decreases angiotensin II induced miR-26a expression in cardiac fibroblasts (Wei et al., 2013). We therefore evaluated whether NF-κB functionally regulated miR-26a expression in IAV-infected cells. To this end, we first confirmed the activation of NF-κB in IAV-infected 293T cells. Accordingly, this activation can be sup- pressed by NF-κB inhibitor Bay11 (Figure 1C–a). We then detected miR-26a expression upon IAV challenge with or without Bay11 treatment. As shown in Figure 1C–b, IAV infection-mediated downregulation of miR-26a can be restored by NF-κB inhibitor Bay11 in 293T cells, suggesting a negative regulatory effect of NF-κB on miR-26a. We also confirmed the inhibitory effect of NF-κB on miR-26a in p65-overexpressed 293T cells (Figure 1C–c). Taken together, these results suggest that IAV infection decreases miR-26a expression mainly through NF-κB signaling pathway.
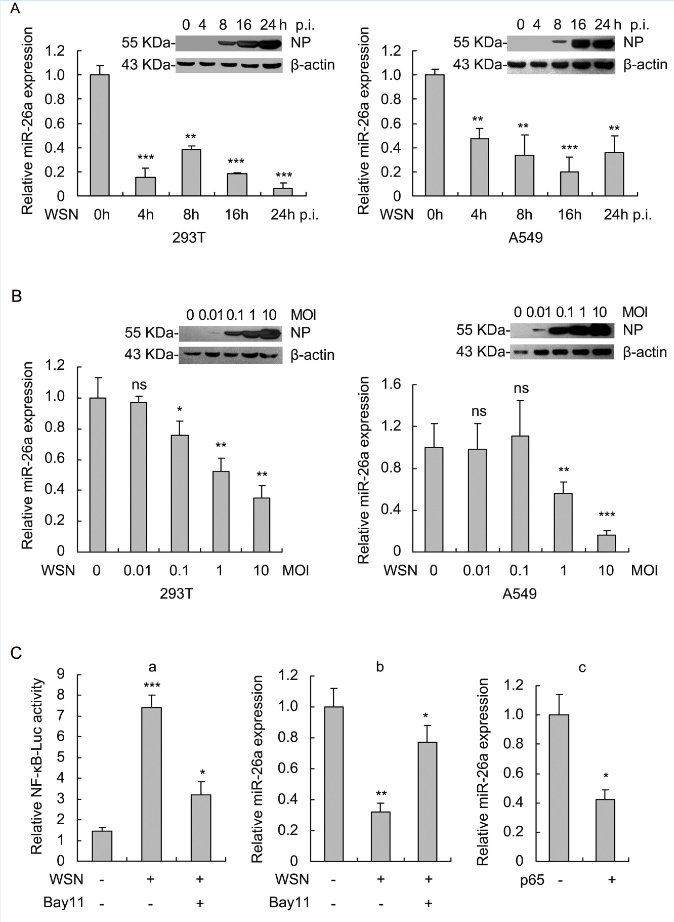
Figure 1. IAV infection induces down-regulation of miR-26a in cells via NF-kB pathway. (A) 293T cells were infected with A/WSN/33 virus at an MOI of 1 and A549 cells were infected at an MOI of 2 for the indicated time. Expression of miR-26a was evaluated by qRT-PCR with U6 as an internal control. NP expression was measured by Western blot to confirm the successful infection and replication of A/WSN/33 virus at 293T and A549 cells. (B) 293T and A549 cells were infected with A/WSN/33 virus at the indicated MOI for 8 h. Expression of miR-26a was evaluated as described in (A). (C) IAV infection down-regulates miR-26a expression mainly through NF-kB pathway. (C-a) 293T cells were transfected with pNF-KB-Luc (400 ng) and pRL-TK (20 ng), which used as an internal control for 24 h followed by infection with A/WSN/33 virus (MOI=1) and incubation with or without NF-kB inhibitor Bay11 for 4 h. At 16 h post-infection, cells were lysed for luciferase activity assay. (C-b) 293T cells were infected with A/WSN/33 virus (MOI=1) followed by treatment with Bay11(10 pmol/L) for 4 h or left untreated. At 8 h post-infection, miR-26a expression was measured as in (A). (C-c) 293T cells were transfected with the plasmid encoding p65 (500 ng) for 24 h and miR-26a expression was measured. Data represent mean ± S.D. of three independent experiments. * P < 0.05, ** P < 0.01, *** P < 0.001; ns, no significant difference
-
To investigate the biological significance of miR-26a during IAV infection, we examined the effect of miR-26a on IAV replication in 293T and A549 cells. To this end, 293T and A549 cells were transfected with miR-26a mimic or miR-26a inhibitor followed by the infection of A/WSN/33 virus at an MOI of 0.5 or 1 and then virus titers in the cultural supernatants were determined by hemagglutinin assay (Transfection with miR-26a shows no cytotoxicity). As shown in Figure 2A, for both 293T and A549 cells, viral titers were decreased significantly when miR-26a was overexpressed while increased when miR-26a was inhibited. Similar results were verified by plaque assay (Figure 2B). We then investigated whether miR-26a inhibits IAV replication in a dose-dependent manner. A549 cells were seeded in a 24-well plate and transfected with miR-26a mimic in increasing dose (12.5, 25, 50, 100, 200 nmol/L) followed by infection with A/WSN/33 virus. At 16 h post-infection, the viral titers in the supernatants were measured by plaque assays. The results in Figure 2C indicated that the cells transfected with a higher dose of miR-26a displayed a stronger antiviral effect. Consistent with the data of viral titers in cultural supernatants, intracellular IAV viral RNA (PB1, NA and M1) replication and viral protein (NP) expression were also decreased in miR-26a-overexpressed cells while increased in miR-26a-inhibited cells (Figure 2D, 2E). Thus, miR-26a functions as a negative regulator of IAV replication.
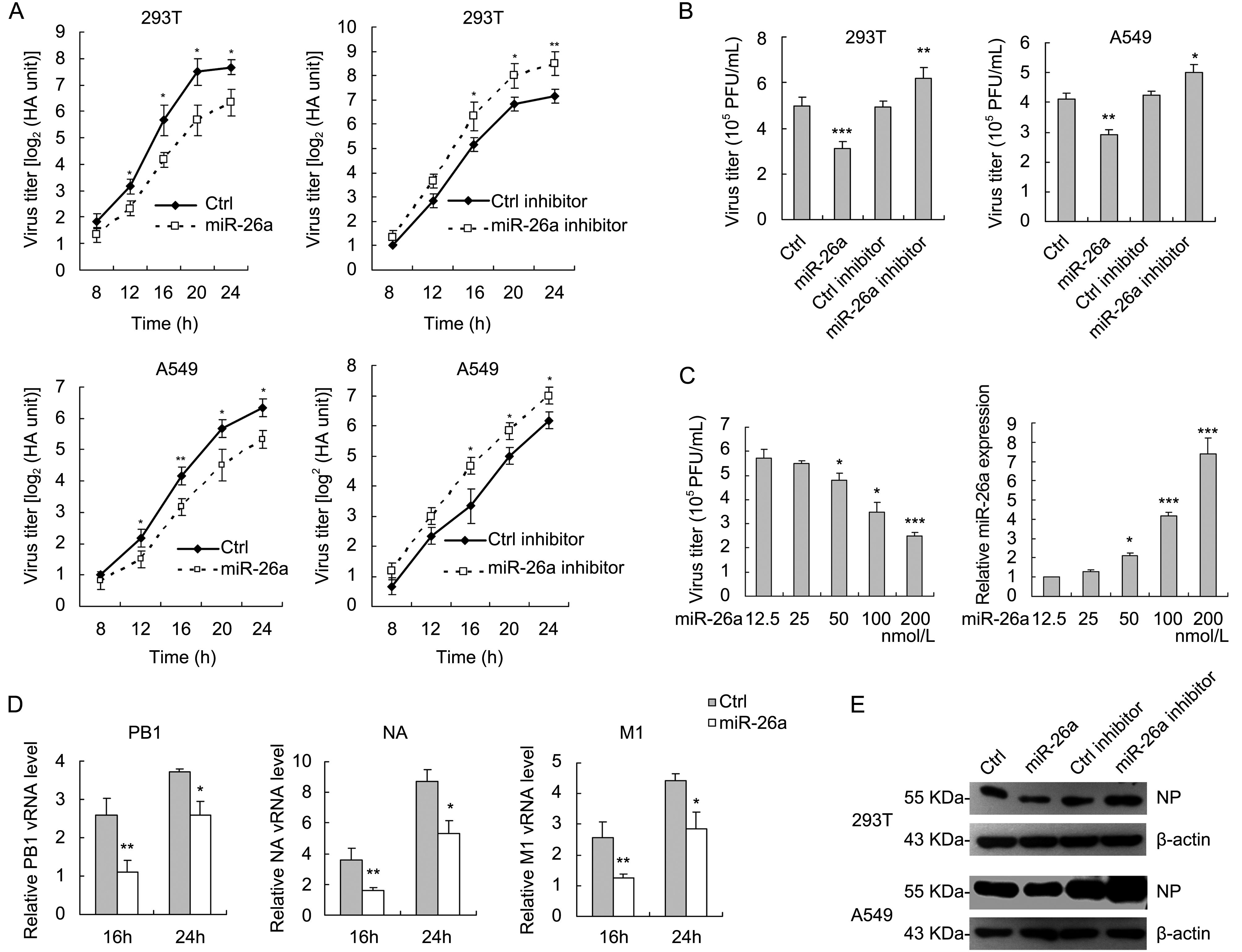
Figure 2. miR-26a attenuates IAV replication. (A) 293T and A549 cells were transfected in a 12-well plate with miR-26a mimic (100 nmol/L) or miR-26a inhibitor (100 nmol/L) for 36 h followed by infection with A/WSN/33 virus at an MOI of 0.5 for 293T cells and MOI of 1 for A549 cells for the indicated time. The supernatants of cell culture were collected and assayed for virus titers by hemagglutinin assay. (B) 293T and A549 cells were transfected with miR-26a mimic (100 nmol/L) or miR-26a inhibitor (100 nmol/L) and infected with A/WSN/33 virus as in (A). At 16 h post-transfection, the media were collected and subjected to plaque assays on MDCK cells. (C) A549 cells were transfected with miR-26a mimic (100 nmol/L) at the indicated concentrations, followed by infection with A/WSN/33 virus at an MOI of 1. At 16 h post-transfection, the supernatants of the cell cultures were harvested and measured by plaque assays as the viral titers and the cell lysates were analyzed by real-time qRT-PCR to confirm the miR-26a mimic transfections. (D) qRT-PCR analysis of viral RNA (PB1, NA, M1) levels in 293T cells transfected with miR-26a mimic (100 nmol/L) infected with A/WSN/33 virus at an MOI of 0.5 for 16 or 24 h. (E) Western blot analysis of NP levels in 293T and A549 cells that transfected with miR-26a mimic (100 nmol/L) or miR-26a inhibitor (100 nmol/L) and infected with A/WSN/33 virus. Data represent mean ± S.D. of three independent experiments. * P < 0.05, ** P < 0.01, *** P < 0.001.
-
To gain an insight into the mechanism of how miR-26a inhibited viral propagation, we first determined whether miR-26a specifically targeted the IAV genome because targeting a specific viral sequence by miRNAs has been shown to be an efficient strategy to inhibit viral replication. To this end, eight fragments of A/WSN/33 genome were inserted into the site of reporter plasmid pRL-TK downstream of the firefly luciferase gene respectively (Figure 3A). The individual reporter vectors (pRL-TK-PB1, pRL-TK-PB2, pRL-TK-PA, pRL-TK-HA, pRL-TK-NA, pRL-TK-NP, pRL-TK-M and pRL-TK-NS) were co-transfected into 293T cells with either miR-26a mimic or mimic control. Luciferase assay was performed at 36 h post transfection and showed no significant difference in relative luciferase activities for the individual vectors containing eight segments of A/ WSN/33 between the cells transfected with miR-26 mimic and mimic control (Figure 3B). Thus, miR-26a did not target IAV genome directly. Further studies to delineate the individual steps (such as viral entry, replication, translation and release) affected by miR-26a may help in understanding how miR-26a regulates IAV replication.
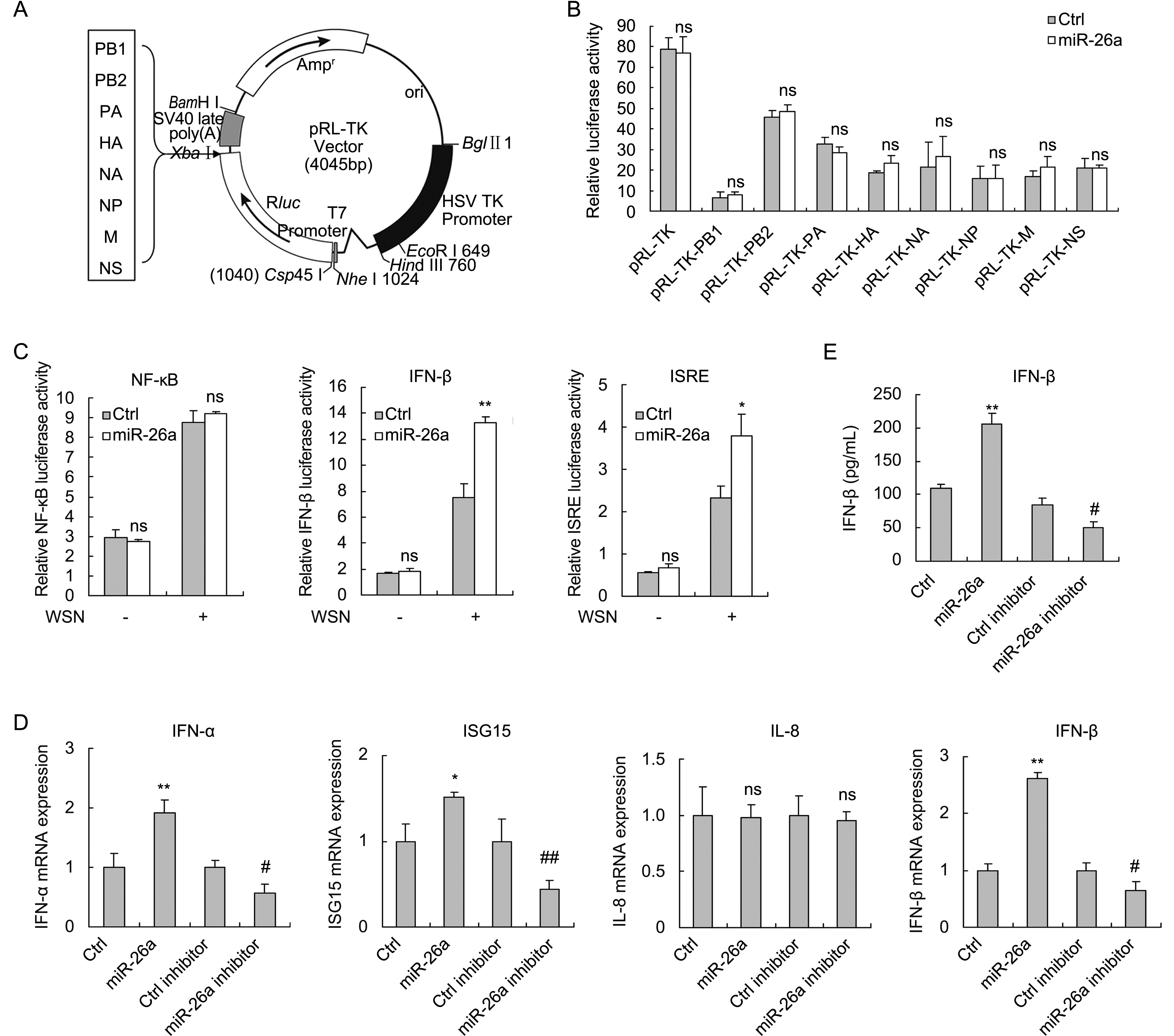
Figure 3. miR-26a does not directly target IAV genome but instead activates IFN signal pathway. (A) Fragments of IAV genome were inserted into the 3′UTR of the luciferase gene in pRL-TK vector as indicated. (B) miR-26a mimic (100 nmol/L) and the constructs (400 ng) were co-transfected into 293T cells. The luciferase activity assays were performed at 36 h post transfection. (C) 293T cells were co-transfected with miR-26a mimic (100 nmol/L), reporter vector pNF-κB-Luc (400 ng), pIFN-β-Luc (400 ng), or pISRE-Luc (400 ng), with pRL-TK (20 ng) as an internal control. At 24 h post-transfection, the cells were infected with A/WSN/33 virus (MOI = 1) or left uninfected. Then the luciferase assays were performed at 12 hpi. (D) 293T cells were transfected with miR-26a mimic (100 nmol/L) or inhibitor (100 nmol/L) followed by infection with A/WSN/33 virus. IFN-α, IFN-β, ISG15 and IL8 mRNA expression were measured by qRT-PCR analysis at 8 hpi. IFN-β levels in the supernatants were measured by ELISA at 16 hpi (E). Data represent mean ± S.D. of three independent experiments.* P < 0.05, ** P < 0.01 compared with the Ctrl group; # P < 0.05, ## P < 0.01 compared with the Ctrl inhibitor group; ns, no significant difference. hpi, hours post infection.
-
Since miR-26a negatively regulated IAV replication but did not target genome of IAV directly, we hypothesized that miR-26a might regulate innate immune system, the first line of defense of a host against virus infection. To verify this hypothesis, luciferase assay was performed in 293T cells transfected with miR-26a and the reporter plasmids (pNF-κB-luc, pIFN-β-luc and pISRE-luc) in the presence or absence of IAV infection. The data showed that IFN-β and ISRE, but not NF-κB promoter activities were enhanced by miR-26a upon IAV infection (Figure 3C). We next examined IFNα/β, IFN-stimulated gene ISG15 and NF-κB-targeted gene IL-8 expression in IAV-infected 293T cells in the presence of miR-26a mimic or miR-26a inhibitor. Real-time PCR analysis showed that overexpression of miR-26a increased IFN-α, IFN-β and ISG15 mRNA levels, while repression of miR-26a decreased IFN-α, IFN-β and ISG15 expression. Furthermore, IFN-β protein levels increased by miR-26a were confirmed by using ELISA assay. However, levels of IL-8, one of the target genes activated by NF-κB, were not changed whether miR-26a was overexpressed or re- pressed (Figure 3D, 3E). Together, these results suggested that miR-26a activates the IFN signaling pathway and promotes the production of IFN-stimulated genes upon IAV infection.
We next investigated what was the major target of miR-26a that could regulate IFN-α/β signaling pathway. 293T cells were co-transfected with miR-26a and luciferase reporter plasmid IFN-α/β-luc, together with several key mediators of IFN signaling pathway, including RIG-I, VISA, TBK1 and IRF3. Dual-luciferase reporter assays showed that miR-26a enhanced the activation of IFN-β promoter induced by RIG-I and VISA, but not that of TBK1 and IRF3 (Figure 4A). The results suggested that the target of miR-26a was upstream of TBK1 in IFN signaling pathway. We then analyzed in silico with TargetScan (www.targetscan.org) to predict the potential targets of miR-26a. 3′-UTR of three negative regulators of IFN signaling act upstream of TBK1 including USP3 (Figure 4B) (Cui et al., 2014), CYLD (Friedman et al., 2008) and TRIM11 (Lee et al., 2013) contains putative binding sites of miR-26a. Luciferase assay showed that miR-26a directly targets USP3 (Figure 4C), but not CYLD or TRIM11 (data not shown). Then we examined whether the protein level of USP3 was targeted and regulated by miR-26a after IAV challenge. As expected, USP3 expression was decreased by miR-26a overexpression and increased by miR-26a inhibition. Thus, miR-26a targets USP3 in IAV-infected cells. To further confirm that miR-26a suppress IAV replication by targeting USP3, we examined the role of USP3 in IAV infection. As shown in Figure 4E, overexpression of USP3 promotes IAV replication in 293T cells. Finally, as mentioned above (Figure 1C-b), IAV infection-mediated downregulation of miR-26a can be restored by NF-κB inhibitor Bay11; we further detected the effect of Bay11 treatment on USP3 protein levels upon IAV infection. As shown in Figure 4F, NF-κB inhibitor Bay11 increased miR-26a expression while decreased USP3 protein levels in IAV-infected 293T cells. This result further confirmed the direct targeting of USP3 by miR-26a upon IAV infection. Taken together, these data show miR-26a triggers IFN responses mainly through directly targeting USP3, a negative regulator of type I IFN signaling pathway, to suppress IAV infection.
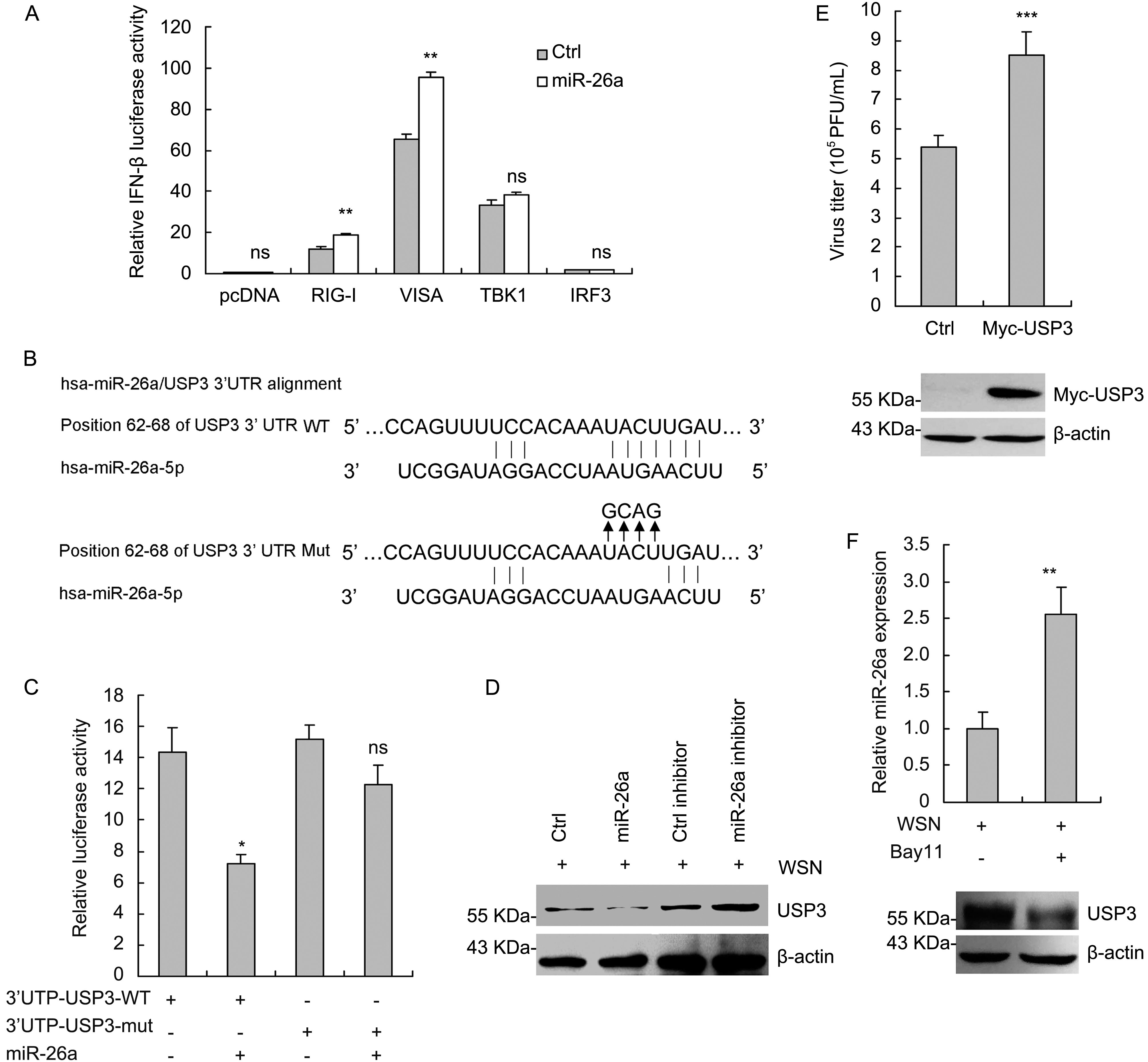
Figure 4. miR-26a targets USP3. (A) 293T cells were co-transfected with the plasmids encoding RIG-I, VISA, TBK1 and IRF3 (200 ng), reporter vector pIFN-β-Luc (200 ng), inter control vector pRL-TK (20 ng) with or without miR-26a mimic (100 nmol/L). At 30 h post-transfection, luciferase activity was measured. (B) Sequence complementarity between miR-26a and the 3′UTR of USP3 gene. (C) 293T cells were transfected with wild type or mutant USP3 3′UTR-luc reporter vector (400 ng), pGL3-control vector (20 ng) along with miR-26a or miR negative control for 36 h. The cells were harvested, and a luciferase assay was performed. (D) USP3 protein levels in 293T cells were analyzed by western blotting after transfection with miR-26a (100 nmol/L) or miR-26a inhibitor (100 nmol/L) for 48 h followed by infection with A/WSN/33 virus (MOI = 1) for another 4 h. (E) 293T cells were transfected with the plasmid encoding Myc-USP3 (500 ng) for 24 h followed by infection with A/WSN/33 virus for another 16 h. The supernatants of the cell cultures were harvested and measured by plaque assays as the viral titers and the cell lysates were analyzed by western blotting to confirm the expression of Myc-USP3. (F) 293T cells were infected with A/WSN/33 virus (MOI = 1) followed by treatment with Bay11 (10 μmol/L) for 4 h or left untreated. miR-26a expression was measured by real-time qRT-PCR and USP3 protein levels were analyzed by western blotting. Data represent mean ± S.D. of three independent experiments. ns, no significant difference.
-
Recently, miR-26a is proved as a typically multifunctional miRNA that involves in many physiological and pathological processes, such as proliferation, innate immunity, neurological diseases and tumorigenesis (Chen et al., 2016). miR-26a has also been identified as one of the significantly downregulated circulating miRNAs in the blood of influenza A H1N1 patients (Tambyah et al., 2013). Furthermore, miR-26a has been reported to repress porcine reproductive and respiratory syndrome virus (PRRSV) replication through by activating innate antiviral IFN pathway but not directly target virus genome (Jia et al., 2015; Li et al., 2015). But the molecular mechanism of the repression of miR-26a expression and the functional role of miR-26a upon IAV replication are not clear. In this study, we show that IAV infection reduces miR-26a expression mainly through NF-κB pathway. Overexpression of miR-26a leads to a significant inhibition of IAV replication. miR-26a does not directly target IAV genome. Instead, miR-26a activates the IFN signaling pathway and promotes the production of IFN-stimulated genes, thereby leading to the suppression of viral replication. Thus, miR-26a is a protective factor against IAV replication. And downregulation of miR-26a may be a new strategy evolved by IAV to counteract cellular antiviral responses.
Upon IAV infection, viral RNAs are recognized by RIG-I, leading to the activation of type I IFN signal pathway and the production of IFN-induced genes. K63-linked ubiquitination of RIG-I plays a critical role in the activation of IFN signaling cascades (Gack et al., 2007). The deubiquitinating enzyme USP3 removes K63-linked polyubiquitin chains from RIG-I to prevent aberrant IFN induction (Cui et al., 2014). Here, we identify miR-26a targets USP3 directly in IAV-infected cells and overexpression of miR-26a promotes the K63-linked ubiquitination of RIG-I whereas has no effect on its K48-linked ubiquitination (Supplementary Figure S1). Thus, miR-26a upregulates innate anti-viral responses by targeting USP3.
miRNA has been shown to be an effective weapon to protect host from virus infection (Baltimore et al., 2008). Viral infection could change miRNA expression profiles. On one hand, these changed miRNAs would trigger innate immune response to inhibit virus replication. On the other hand, the change of cellular miRNA can be induced by specific virus to create a cellular environment appropriate for virus replication. In this study, miR-26a inhibits IAV replication through triggering IFN pathway. IAV infection reduces expression of miR-26a, which created an environment facilitating IAV replication. Our results indicated that miR-26a might be an effective measure to cure and prevent IAVs.
-
This work was supported by grants from the National Basic Research Program of China (973 Program, No. 2012CB518900) and the Beijing Natural Science Foundation (No. 7122109).
-
The authors declare that they have no conflict of interest. This article does not contain any studies with human or animal subjects performed by any of the authors.
-
GSJ, SLP, and HWL designed the study. GSJ, LJD, SLP, and WJX carried out the experiments. GSJ, LJD, SLP, WJX, and HWL analyzed the data. GSJ and LJD wrote the paper. All authors have read and approved the final manuscript
Supplementary Figure S1 is available on the websites of Virologica Sinica: www.virosin.org; link.springer.com/journal/12250.
-
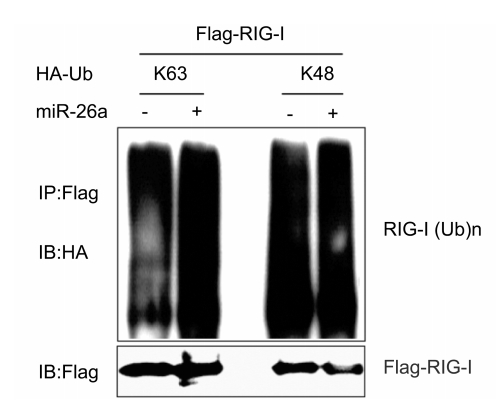
Figure S1. miR-26a promotes the K63-linked ubiquitination of RIG-I. Lysates of 293T cells transfected with plasmids for Flag-RIG-I and HA-tagged K63-linked ubiquitin (HA-K63-Ub) or HA-tagged K48-linked ubiquitin (HA-K48-Ub), together with miR-26a or its negative control were immunoprecipitated with anti-Flag and immunoblotted with anti-HA
Influenza A virus-induced downregulation of miR-26a contributes to reduced IFNα/β production
-
Shijuan Gao
1,#,,
 ,
, - Jiandong Li 2,# ,
- Liping Song 4 ,
- Jiaoxiang Wu 2 ,
-
Wenlin Huang
2,3,,
- Received Date: 22 April 2017
- Accepted Date: 21 June 2017
- Published Date: 30 June 2017
Abstract: Innate immunity provides immediate defense against viral infection.Influenza A virus (IAV) is able to get past the first line of defense.Elucidation of the molecular interaction between influenza factors and the newly recognized host players in the innate response might help in our understanding of the root causes of virulence and pathogenicity of IAV.In this study,we show that expression of miR-26a leads to a significant inhibition of IAV replication.miR-26a does not directly target IAV genome.Instead,miR-26a activates the type I interferon (IFN) signaling pathway and promotes the production of IFN-stimulated genes,thus suppressing viral replication.Furthermore, ubiquitin-specific protease 3(USP3),a negative regulator of type I IFN pathway,is targeted by miR- 26a upon IAV challenge.However,miR-26a is significantly downregulated during IAV infection. Thus,downregulation of miR-26a is a new strategy evolved by IAV to counteract cellular antiviral responses.Our findings indicate that delivery of miR-26a may be a potential strategy for anti-IAV therapies.

 DownLoad:
DownLoad: