









-
Kaposi’s sarcoma-associated herpesvirus (KSHV, also known as human herpes virus-8, HHV8) is the causative agent for a number of cancers, including Kaposi’s sarcoma (KS), primary effusion lymphoma (PEL) and multicentric Castleman disease (MCD), all of which arise preferentially in immunocompromised patients (Chang et al., 1994; Cesarman et al., 1995; Soulier et al., 1995). Currently, there are four KS isoforms: classic KS affecting elderly men of the Mediterranean; endemic KS, existing in some countries of Central and Eastern Africa; iatrogenic KS, which usually develops in organ transplant recipients with immunosuppression; and epidemic or AIDS-KS, which typically presents with more aggressive features (Mesri et al., 2010). Even though the combined antiretroviral therapy (cART) helps to reduce the total incidence of KS in the western world, KS is still the most common AIDS-associated malignancy and a leading cause of cancer-related morbidity and mortality in AIDS patients (Bonnet et al., 2004). PEL is a B-cell malignancy harboring KSHV which arises preferentially within the pleural or peritoneal cavities of immunocompromised patients (Cesarman et al., 1995). PEL is a rapidly progressing malignancy with a median survival time of approximately 6 months, even after the combinational chemotherapy (Chen et al., 2007). KSHV-associated MCD is a rare lymphoproliferative disorder that frequently arises in HIV+ patients who have a suppressed HIV activity and a relatively preserved CD4 count (Wang et al., 2016). Like other herpesviruses, KSHV establishes a lifelong infection in the host utilizing two major distinct phases: latent infection and lytic replication. During the latent infection—the predominant phase in the majority of infected cells—only a limited number of viral genes are expressed. Provocation by a variety of stimuli induces lytic replication, resulting in new virion assembly and release of infectious viral particles (Schulz, 2006). Previous studies suggest that the oncogenic potential of KSHV is largely dependent upon genes expressed during the viral latency, however, recent data demonstrate that the viral lytic reactivation is critical for infection of naïve cell targets, maintenance of the KSHV reservoir, and tumor development (Aluigi et al., 1996; Lebbe et al., 1997; Grundhoff and Ganem, 2014;).
Lipids form a diverse group of water-insoluble molecules that include triacylglycerides, phosphoglycerides, sterols and sphingolipids. Lipids are essential for mammalian cells to maintain their physiological functions. For instance, fatty acids are the major building blocks for the synthesis of triacylglycerides during the process of energy storage. Phosphoglycerides, together with sterols and sphingolipids are the major structural components of cellular membranes. In addition, lipids as second messengers and hormones also play important roles in many signal transduction pathways. For example, lipids in the cellular membranes have been linked to the functions of several signal transduction pathways, including immunoglobulin E signaling (Sheets et al., 1999), T-cell antigen receptor signaling (Janes et al., 2000), glial-cell-derived neurotrophic factor (GDNF) signaling (Tansey et al., 2000), Ras signaling (Roy et al., 1999) and Hedgehog signaling (Porter et al., 1996). Accumulating evidence has shown that cancer cells develop specific alterations in different aspects of lipid metabolism to facilitate their survival and various malignant behaviors. To date, there are only a handful of studies describing how an oncogenic virus such as KSHV can manipulate host cellular lipid biosynthesis and metabolism to promote viral infection, pathogenesis, and tumorigenesis. In the current review, we summarize recent findings in this new area of KSHV research.
-
In cell culture, KSHV is able to infect various types of human cells, such as B cells, endothelial cells, epithelial cells, and fibroblasts (Dai et al., 2012; Fontana et al., 2014; Kang and Myoung, 2017). Several membrane proteins including heparin sulfate proteoglycan (HSPG), DC-SIGN, integrin α3β1/αvβ3, EphA2 and xCT can act as cellular receptors for KSHV infection in a cell type-dependent manner (Birkmann et al., 2001; Akula et al., 2002; Kaleeba and Berger, 2006; Rappocciolo et al., 2006; Garrigues et al., 2008; Hahn et al., 2012). After binding with these receptors, KSHV can induce the phosphorylation of focal adhesion kinase (FAK) which subsequently leads to the activation of Src, phosphatidylinositol 3-kinase (PI3-K), protein kinase C-ζ (PKC-ζ), Rho-GTPases, mitogen-activated protein kinase kinase (MEK), and extracellular signal regulated kinase 1/2 (ERK1/2) (Naranatt et al., 2003; Sharma-Walia et al., 2004; 2005). Activation of these signaling cascades can facilitate virus entry, its movement in the cytoplasm, and the nuclear delivery of viral DNA. Many of these KSHV induced signaling molecules are associated with lipid rafts micro-domains in the membrane. Previously, Raghu et al. reported that lipid rafts of endothelial cells play critical roles in KSHV infection and gene expression (Raghu et al., 2007). They found that disruption of lipid rafts by methyl β-cyclo dextrin (MβCD) or nystatin significantly inhibited the expression of viral latent gene, Lana (Latency-associated nuclear antigen), and the lytic gene, Rta (Replication and transcription activator). Lana is the only viral protein consistently expressed in all KS-associated malignancies (Dupin et al., 1999) and its major function is to maintain the viral episome in the latently-infected cells (Ballestas et al., 1999; Avey et al., 2015). Rta is a key viral protein initially controlling virus “latent to lytic” switch (Sun et al., 1998). The inhibition of Lana and Rta expression was mainly achieved by suppressing the KSHV-induced PI3-K and RhoA-GTPases activation and reducing the co-localizations of PI3-K and RhoA-GTPases with lipid rafts (Raghu et al., 2007). Since disruption of lipid rafts did not affect KSHV binding and viral DNA internalization, the authors concluded that lipid rafts are mainly required for KSHV-induced microtubule dynamics, virus movement in the cytoplasm, nuclear delivery of viral DNA, and viral gene expression (Raghu et al., 2007). A later study from the same group indicates that at a very early time-point during infection (~1 min post-infection), an adaptor protein, c-Cbl, can induce the selective translocation of KSHV into the lipid rafts along with the α3β1, αVβ3, and x-CT receptors, leading to a productive infection (Chakraborty et al., 2011). Knock-down of c-Cbl was found to inhibit KSHV infection by preventing micropinocytosis and selective virus-receptor translocation, with KSHV being diverted toward a clathrin-lysosomal noninfectious pathway.
One recent study has shown that KSHV infection can activate several components of the lipoxygenase pathway, including 5-lipoxygenase (5LO), leukotriene (LT) A4 hylase (LTA4H), and leukotriene B4 (LTB4), a chemotactic lipid mediator of the 5LO pathway (Sharma-Walia et al., 2014). Interestingly, blocking the 5LO/LTB4 cascade can inhibit the expression of KSHV-encoded latent protein Lana, the immunomodulatory protein K5, the viral macrophage inflammatory protein 1 (MIP-1), and MIP-2 expression. Taken together, these results clearly indicate that cellular lipids, lipid metabolism and related signaling pathways are involved in KSHV primary infection and subsequent latency establishment. Given its critical role in KSHV infection cycle, the lipid pathway may represent a promising “drug target” to manage KSHV infection.
-
Like latency, viral reactivation and lytic replication also play important roles in KSHV oncogenesis. A recent study has shown that reactivation can be induced by some short-chain fatty acids (SCFAs) such as phenylbutyrate through inhibiting histone deacetylase (HDAC) activities (Gorres et al., 2014). Consistently, Yu et al. have found that several SCFAs produced by periodontal pathogens such as Porphyromonas gingivalis and Fusobacterium nucleatum can also induce the KSHV lytic reactivation by suppressing HDACs as well as two histone N-lysine methyltransferases (HLMTs): enhancer of zeste homolog2 (EZH2) and suppressor of variegation 3–9 homolog1 (SUV39H1) (Yu et al., 2014). These findings indicate that periodontal pathogens may create a unique microenvironment in the oral cavity, which in turns favors KSHV replication and KS development. Indeed, oral cavity involvement represents the initial manifestation of KS in 20%–60% of HIV-associated cases (Flaitz et al., 1997; Lager et al., 2003; Reichart 2003).
We recently reported that targeting sphingolipid metabolism by either sphingosine kinase inhibitors or exogenous ceramides can dramatically induce viral lytic genes expression in KSHV-infected primary endothelial cells or PEL cells (Qin et al., 2014; Dai et al., 2014, 2015). Such induction is at least in part mediated by the suppression of pro-latency viral microRNAs (e.g., miR-K12-1 and miR-K12-11) as well as related signaling pathways (e.g., NF-κB) (Dai et al., 2014).
-
Recent studies have shown that cellular lipids and lipid metabolism can regulate the survival of KSHV-infected primary and tumor cells. Having analyzed the metabolic profiles of primary B cells and KSHV+ PEL cells, Bhatt et al. found that KSHV+ PEL cells exhibit greater aerobic glycolysis and fatty acid synthesis than primary B cells (Bhatt et al., 2012). Meanwhile, the major lipid components of eukaryotic cell walls (e.g., phosphatidylcholine and phosphatidylethanolamine) are also more abundant in PEL cells. The fatty acid synthase (FASN), a multi-enzyme complex involved in the cellular lipids synthesis (Kuhajda et al., 2000), is overexpressed in PEL cells. Moreover, treatment of KSHV+ PEL cells with the FASN inhibitor, C75, can reduce cell viability in a dose-dependent manner (Bhatt et al., 2012).
Delgado et al. have utilized a metabolomic approach to investigate the KSHV mediated global metabolic alterations in latently infected cells (Delgado et al., 2012). They found that ~60 analyzed metabolites were altered after latent infection. Among them, many long chain fatty acids were affected due to the alteration of fatty acid synthesis pathways. Previous studies have shown that fatty acid synthesis is also required for the survival of latently infected endothelial cells and inhibition of key enzymes (e.g., acetyl-CoA carboxylase (Wang et al., 2009) or FASN (Kuhajda et al., 2000)) in this pathway led to apoptosis of infected cells. In contrast, addition of palmitic acid (the fundamental fatty acid precursor) can protect latently infected cells from the acetyl-CoA carboxylase inhibitor, 5-(Tetradecyloxy)-2-Furoic Acid (TOFA)-induced cell death. The same group later reported that the KSHV latent infection also increases peroxisome biogenesis. Interestingly, the proteins involved in peroxisomal lipid metabolism of very long chain fatty acids, such as ABCD3 (a peroxisome-specific lipid transporter) and ACOX1 (Acyl-CoA Oxidase 1, a peroxisomal enzyme), are required for the survival of latently infected cells (Sychev et al., 2017).
Sphingolipid biosynthesis involves hydrolytic conversion of ceramide to sphingosine. Subsequently, sphingosine is phosphorylated by one of two sphingosine kinase isoforms (SphK1 or SphK2) to generate bioactive sphingosine-1-phosphate (S1P) (Ogretmen and Hannun, 2004) (Figure 1). The relative levels of ceramide and S1P ultimately determine the fate of tumor cells, with accumulation of ceramides favoring apoptosis, and accumulation of S1P favoring proliferation (Cuvillier et al., 1996; Ogretmen and Hannun, 2004). SphK can be activated by a variety of tumor-promoting cytokines and growth factors. SphK activation is responsible for a rapid accumulation of intracellular S1P and depletion of ceramide species (Maceyka et al., 2002). S1P can subsequently bind to one of five G protein-coupled S1P receptors (S1PR1-5) and then activate diverse downstream signaling pathways (Strub et al., 2010). Because of their pleiotropic roles, bioactive sphingolipids have evolved as promising therapeutic targets for cancer treatment over the past two decades (Saddoughi et al., 2013). We have recently reported that induction of intracellular ceramide using a novel SphK2 inhibitor (ABC294640) or exogenous ceramide/dihydro(dh)-ceramide species (e.g., C6-Cer or dhC16-Cer) can effectively kill KSHV+ primary endothelial cells or PEL tumor cells, but have little effect on KSHV non-infected cell controls (e.g., naïve endothelial cells or B cells) (Qin et al., 2014; Dai et al., 2014, 2015). Further, these compounds can also repress KSHV+ PEL tumor progression in vivo and it is likely that this is mediated through interfering with several cell survival/proliferation-associated signaling pathways (e.g., MAPK/ERK, Akt and NF-κB) and up-regulating viral lytic genes and cellular tumor suppressor genes expression (Qin et al., 2014; Dai et al., 2015; Cao et al., 2017). In addition, the KSHV mediated up-regulation of SphK2 (Dai et al., 2014) may also help sensitize KSHV+ cells to sphingolipid targeted therapy.

Figure 1. Targeting sphingolipid metabolism in KSHV-infected host cells. CerS, ceramide synthase; S1PP, S1P phosphatase; SphKs, sphingosine kinases; S1PRs, S1P receptors. ABC294640: A novel selective SPHK2 inhibitor; C6-Cer, dhC16-Cer etc: exogenous short- or long-chain ceramides.
-
One recent study revealed that neutral lipid (NL) content is increased in KSHV-infected human umbilical vein endothelial cells (HUVEC) (Angius et al., 2015). In particular, triglyceride synthesis is boosted in the lytic phase, whereas the cholesteryl ester synthesis rises in the latent phase. Moreover, inhibition of cholesterol esterification significantly reduces neo-tubule formation mainly in latently infected cells, indicating that a reprogramming of cholesteryl ester metabolism is involved in KSHV-mediated neo-angiogenesis and that it may also contribute to the high metastatic potential of the derived-tumors.
It is believed that KSHV-encoded G protein-coupled receptor (vGPCR) is a key molecule in the pathogenesis of KS and that it plays a central role in promoting vascular endothelial growth factor-driven angiogenesis and spindle cell proliferation (Montaner et al., 2003; Grisotto et al., 2006; Wei et al., 2016). Several studies have shown that 1 Alpha, 25-dihydroxyvitamin D3 [1 alpha, 25(OH)(2)D(3)] and its TX527 analog inhibit the growth of vGPCR transformed endothelial cells in vitro and in vivo. The inhibition effects are achieved through a complex of mechanisms including an interaction with vitamin D receptor, down-regulation of the NF-κB pathway and up-regulation of the pro-apoptotic protein, Bim (Gonzalez-Pardo et al., 2010; 2012; 2013; Suares et al., 2015). Taken together, these data indicate the importance of vitamin D as a steroid signaling molecule in vGPCR-transformed endothelial cell proliferation.
-
Our group and others have recently shown that cellular lipids and lipid metabolism play important roles in KSHV-infected cell survival, pathogenesis, and tumorigenesis (summarized in Figure 2). Lipid research has become an exciting direction in the KSHV field. To date, it is still largely unclear how this oncogenic virus manipulates lipid biosynthesis and metabolism during de novo infection and KSHV mediated tumor development. Clinically, there are few data resulting from clinical trials testing the effectiveness of lipids-targeted therapeutics for KSHV-related malignancies. To the best of our knowledge, there’s only one ongoing early phase trial for the evaluation of ABC294640 in patients with refractory/relapsed Diffuse Large B-cell Lymphoma (DLBCL) or Kaposi Sarcoma (KS) directed by Dr. Suki Subbiah (NCT02229981). How to accelerate the “bench to bedside” transition in this field is a key question needs to be addressed soon.
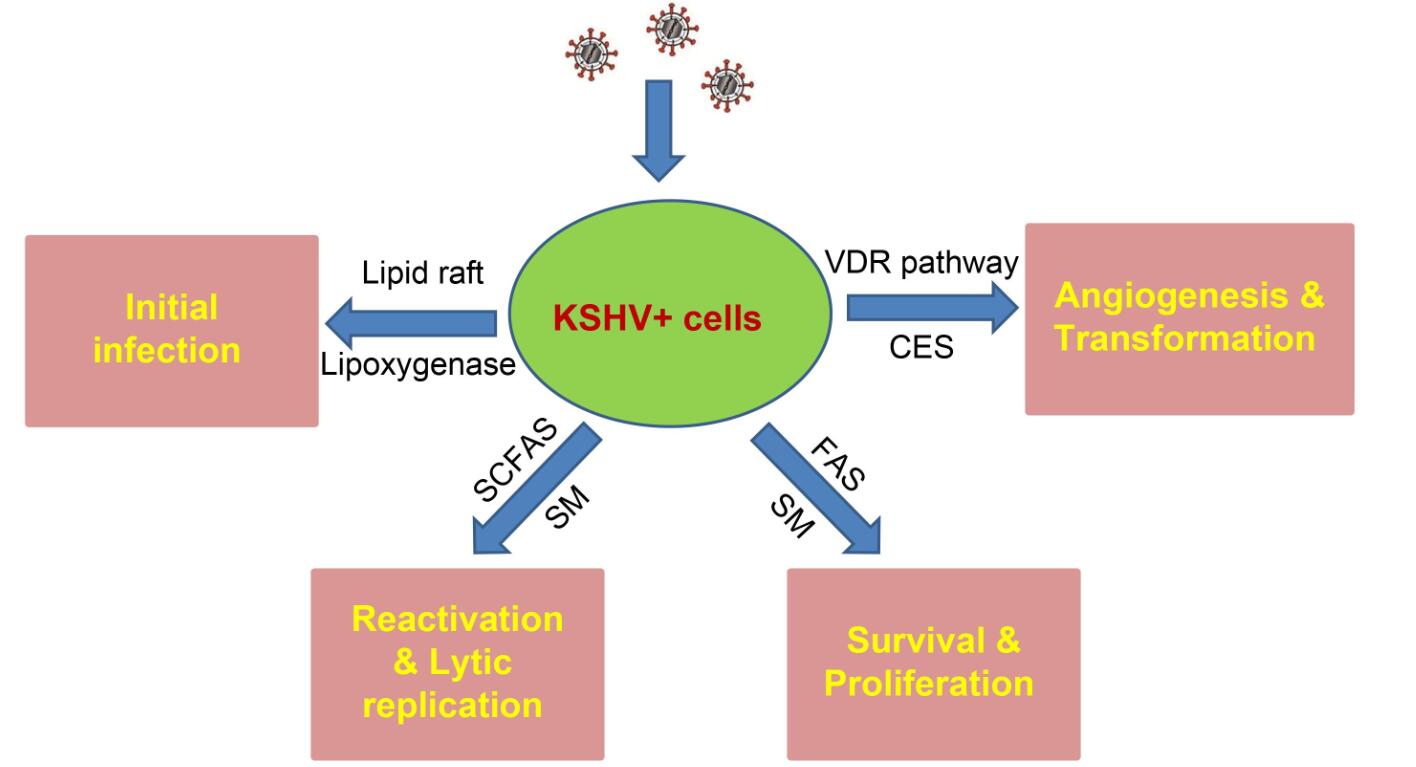
Figure 2. Schematic of recent findings about the contributions of cellular lipids and lipid metabolism to KSHV infection and pathogenesis. SCFAs: short-chain fatty acids; SM: sphingolipid metabolism; FAS: fatty acid synthesis; VDR: Vitamin D receptor; CES: cholesteryl ester synthesis.
-
This work was partially supported by grants from a DOD Career Development Award (CA140437), the Louisiana Clinical and Translational Science Center Pilot grants (U54GM104940 from NIH), a LSU LIFT2 funding, a NIH P20-GM121288-01 subproject, NIH RO1s (AI091526, AI128864, AI101046, and AI106676) as well as awards from the National Natural Science Foundation of China (81472547, 81400164, 81672924 and 81772930). Funding sources had no role in the study design, data collection/analysis, decision to publish, and/or manuscript preparation.
-
The authors declare that they have no conflicts of interest. This article does not contain any studies with human or animal subjects performed by any of the authors.
Lipids, lipid metabolism and Kaposi’s sarcoma-associated herpesvirus pathogenesis
-
Lu Dai
1,2,3,,
 ,
, - Zhen Lin 4 ,
- Wei Jiang 5 ,
- Erik K. Flemington 4 ,
-
Zhiqiang Qin
1,2,3,,
- Received Date: 02 June 2017
- Accepted Date: 05 September 2017
- Published Date: 10 October 2017
Abstract: Lipids are essential for mammalian cells to maintain many physiological functions. Emerging evidence has shown that cancer cells can develop specific alterations in lipid biosynthesis and metabolism to facilitate their survival and various malignant behaviors. To date, the precise role of cellular lipids and lipid metabolism in viral oncogenesis is still largely unclear with only a handful of literature covering this topic to implicate lipid metabolism in oncogenic virus associated pathogenesis. In this review, we focus on the role of lipid biosynthesis and metabolism in the pathogenesis of the Kaposi’s sarcoma-associated herpesvirus, a common causative factor for cancers arising in the immunocompromised settings.

 DownLoad:
DownLoad: