









-
Hepatitis C virus (HCV) is an enveloped, single-stranded positive-sense RNA virus belonging to the genus Hepacivirus of the Flaviviridae family (Choo et al., 1989). Upon attaching to specific cell surface receptors on hepatocytes, the viral/receptor complex is then internalized. The 9.6 kb HCV viral genome encodes a polyprotein precursor, which is translated and processed to generate at least 10 structural and nonstructural proteins (Sakamoto and Watanabe, 2009). All of these proteins are important in the life-cycle of HCV. Establishment of the HCV subgenomic replication system (Con1) in 1999 (Lohmann et al., 1999) and the cell model for infection (HCVcc) in 2005 has greatly accelerated studies of HCV (Wakita et al., 2005). Major developments have been made during the last 20 years. However, HCV biology remains incompletely understood, particularly with regard to the interface between the virus and cell host. In addition to viral-encoded proteins, a myriad of cellular proteins have been shown to either promote or inhibit HCV propagation (Zhong et al., 2005; Li et al., 2014).
The normal cellular prion protein (PrP) is a glycophosphatidylinositol (GPI)-anchored glycoprotein composed of 254 amino acids (aa). The first 22 aa at the N-terminus comprise the leader peptide, guiding PrP entry into the endoplasmic reticulum (ER) to be cleaved. The last 23 aa at the C-terminus comprise the GPI-peptide signaling sequence (GPI-PSS), which is replaced by a fully assembled GPI anchor. After glycosylation at N181 and/or N197, PrP in most cell types is secreted via the trans-Golgi network and then tethered in lipid rafts with its GPI anchor (Yang X et al., 2014). However, in some cancer cells, due to dysfunction of the GPI synthesis machinery, PrP is expressed as pro-PrP, defined as having retained its GPI-PSS. Importantly, in the GPI-PSS of PrP, there is a filamin A (FLNa)-binding motif enabling pro-PrP binding. FLNa is a linker protein that mediates the interaction between cell surface receptors and the actin cytoskeleton. Thus, unlike GPI-anchored PrP, which is found mostly on the outer cell membrane, some pro-PrP also localizes inside the cytosol (Li et al., 2009; Yang et al., 2016). More recent studies have revealed that failure to remove the GPI-PSS is due to multiple deficiencies in the GPI anchor modification machinery (Li et al., 2009; Yang et al., 2016).
Although PrP is highly conserved evolutionarily among mammals and widely expressed in many tissues, particularly the central nervous system (CNS) and lymph-reticular system, its physiological function remains unclear. PrP-null mice, cattle, and goats develop normally without obvious defects (Prusiner et al., 1993; Sailer et al., 1994; Richt et al., 2007; Benestad et al., 2012). However, PrP has been shown to have several important roles in cell models, including metal homeostasis, cell adhesion and migration, and signal transduction (Brown et al., 1997; Kuwahara et al., 1999; Mouillet-Richard et al., 2000; Bounhar et al., 2001; Paitel et al., 2003; Gao et al., 2016). Interestingly, several reports have suggested that PrP possesses antiviral properties, exhibiting activity against Coxsackievirus B3, endogenous murine retrovirus, murine leukemia virus, adenovirus 5, and human immunodeficiency virus type 1 (HIV-1) (Nakamura et al., 2003; Nikles et al., 2005; Lötscher et al., 2007; Caruso et al., 2009). Although the antiviral mechanism of PrP against most reported viruses is unknown, one study suggested that PrP binds to viral genomic RNA to suppress HIV RNA translation (Alais et al., 2012).
In this study, we examined the expression and distribution of PrP in Huh7-Con1 and Lunet cells. In addition, we evaluated the effects of overexpression of wild-type (WT) PrP and PrP harboring a KKRPK motif deletion on the expression of HCV proteins. Our findings provided important insights into the role of PrP in inhibiting HCV replication.
-
Huh7-Con1 and Lunet human hepatocellular carcinoma cells were kindly provided by Professor Xinwen Chen (Wuhan Institute of Virology, Chinese Academy of Sciences). Lunet cells were derived from Huh7 cell and generated by curing Huh7 replicon cells with a selective drug (Friebe et al., 2005; Koutsoudakis et al., 2007). The cells were cultured in Dulbecco’s modified Eagle medium (11965-092, Gibco, New York, USA) supplemented with 10% fetal bovine serum (FBS, 10099-141, Gibco), 100 U/mL penicillin, and 100 μg/mL streptomycin (PS) in 95% humidity with 5% CO2 at 37 °C. Huh7-Con1 cells (Con1, HCV genotype 1b) harboring the subgenomic HCV replicon (pFKI389neo/NS3-3') were derived from Huh7 cells transfected with the HCV replicon and selected with G418 (Lohmann et al., 1999). Huh7-Con1 cells were cultured in the same medium as Lunet cells with the addition of 0.5 mg/mL G418 (345810; Merck Millipore; CA; USA) (Blight et al., 2002). WVT human neuroblastoma cells expressing PrP were cultured in RPMI 1640 (31800-022, Life Technologies, CA, USA) supplemented with 1.5 g/L sodium bicarbonate, 10% FBS, 1% sodium pyruvate, 1 mmol/L HEPES, 4.5 g/L glucose, glutamine, and PS. The cell lines used in this article were free of mycoplasma contamination.
-
Lentivirus vector pHAGE-CMV-MCS-IZsGreen (pHAGE) and the packaging plasmids pMD2.G and psPAX2 were kindly provided by Professor Cunyi Zheng (Wuhan University, China). The WT human PrP expression vector pcDNA3.1-PrP (1–254) was constructed as previously described (Liu et al., 2002). The entire PrP sequence was removed from pcDNA3.1 with restriction enzymes Xba I and BamH I and inserted into pHAGE. The expression vectors for mutant human PrP (PrPΔ23–27, PrPΔ97–102, and PrPΔ226–231) were constructed using a site-directed gene mutagenesis kit (D0206, Beyotime, China) from pcDNA3.1-PrP. The primer pairs used for amplification are listed in Supplementary Table S1. The sequences of all the constructs were confirmed.
Anti-PrP monoclonal antibodies (mAbs) 4H2, 8B4, and 8H4 were produced, purified, and characterized as previously reported (Zanusso et al., 1998; Yang L et al., 2014). Abs against HCV NS3 (ab65407) were purchased from Abcam (Cambridge, UK). NS5B (sc57800) was purchased from Santa Cruz Biotechnology (Dallas, TX, USA). Mouse anti-actin Abs (KM8001) were purchased from Tianjin Sungene Biotech (Tianjin, China). Mouse IgG1 and rabbit IgG isotype controls were purchased from Biolegend (San Diego, CA, USA). Horseradish peroxidase (HRP)-conjugated goat anti-mouse IgG (7076) was purchased from Cell Signaling Technology (MA, USA). 4′, 6-Diamidine-2′-phenylindole dihydro-chloride (DAPI) was purchased from Roche (10236276001, Mannheim, Germany). Alexa Fluor488 dye-linked goat anti-mouse secondary Abs (A-11001) and Alexa Fluor555 dye-linked goat anti-rabbit IgG (A-21429) were purchased from Invitrogen Corporation (Carlsbad, CA, USA). All the other reagents purchased from commercial sources were used according to the suppliers’ recommendations. Immobilon Western HRP substrate (WBKLS0500) was purchased from Millipore (MA, USA). WesternBright Sirius HRP substrate (K-1121043-D20) was purchased from Advansta (CA, USA).
-
Cultured cells were harvested and lysed as previously described (Li et al., 2009). The concentration of proteins was measured using a Bio-Rad Protein Assay kit (500-0002; Bio-Rad, Hercules, CA, USA). Samples were mixed with 4 × sodium dodecyl sulfate (SDS) loading buffer and heated at 100 °C for 5 min before separation by SDS-polyacrylamide gel electrophoresis (SDS-PAGE) on 10% gels. The proteins were then transferred to nitrocellulose membranes (9004700, Billerica, MA, USA). After blocking in 3% bovine serum albumin (BSA) in Tris-buffered saline (TBS) with 0.1% Tween-20, the separated proteins were incubated with specific primary Abs and then probed with corresponding HRP-conjugated secondary Abs. The concentration of anti-PrP Abs 4H2, 8B4, and 8H4 was 1 μg/mL. All the other primary Abs were used according to the manufacturer’s instruction.
-
Cells were seeded on confocal Petri dishes (801002, NEST, China) for 12 h before staining. The cells were then washed with 0.05% phosphate-buffered saline with Tween-20 (PBST) three times and fixed in 4% (w/v) paraformaldehyde for 15 min at 25 °C. After washing with 0.05% PBST three times, the cells were blocked with 10% goat serum plus 1% BSA in PBST at 25 °C for 1 h and incubated with primary Abs for 1 h at 25 °C. After washing six times, the cells were detected with Alexa Fluor-conjugated secondary Abs in the dark for 1 h at 25 °C. Nuclei were counter-stained with 500 ng/mL DAPI for 3 min. Images were taken with an UltraView Vox confocal microscope (Perkin Elmer) after adding antifade mounting medium (P0128; Beyotime).
-
Total RNA was extracted using an RNApure Tissue Kit (CW0584, CW Biotech, China). One microgram of total RNA was used to synthesize first-strand cDNA using a PrimeScript RT reagent kit (DRR047A, Takara Bio, Japan). qPCR amplification of PrP and HCV was carried out using iQTM SYBR® Green Supermix with primer pairs listed in Supplementary Table S1 (170-8882AP, Bio-Rad). The quantification of PrP and HCV was normalized to the mRNA level of glyceraldehyde 3-phosphate dehydrogenase.
-
Cells were seeded on 6-cm Petri dishes for 12 h and then treated with 0.25% trypsin/ethylenediaminetetraacetic acid. Cell suspensions were centrifuged at 300 × g for 3 min at 4 °C. The collected cell pellets were washed twice with ice-cold PBS and then incubated with PI-PLC (0.1 U/mL, P5542, Sigma) at 37 °C for 1 h. After washing twice with PBS, the cells were incubated with anti-PrP Abs (8H4) or mouse IgG1 isotype control (10 μg/mL) diluted in PBS for 30 min at 4 °C. Bound Abs were detected with APC-conjugated secondary Abs (2 μg/mL; 405308; Bio-legend) for 30 min at 4 °C and analyzed using a BD FACS C6 flow cytometer (model 648282, San Jose, CA, USA).
-
WT and mutant PrP were inserted into the pHAGE vector and transfected into 293T cells together with the packaging plasmids pMD2.G and psPAX2 to generate lentiviral particles as previously described (Li et al., 2009). Lentiviral particles were collected by centrifuging at 600 × g for 10 min to obtain viral supernatants and filtered with a 0.45-μm filter. Huh7-Con1 cells were transduced with the virions for 6 h in the presence of 7.5 μg/mL polybrene (107689; Sigma, USA). PrP expression was confirmed by qPCR and immunoblotting analysis. Virion particles without PrP were included as a negative control.
-
The experiments were analyzed using Student’s t-tests and repeated three times. Statistical graphs were created using GraphPad Prism software. Quantitative data were expressed as standard errors of the means (SEMs). Differences with P values of less than 0.05 were considered statistically significant.
-
Although viral infection has been shown to upregulate the expression of PrP (Müller et al., 1992; Lötscher et al., 2007), whether PrP expression is enhanced in HCV infection is still unclear. Thus, we investigated whether PrP expression was upregulated in Huh7-Con1 HCV subgenomic replicon cells compared with that in Lunet cells, which do not permit HCV replication. We found that there was significant upregulation of PrP mRNA in Huh7-Con1 cells (Con1) compared with Lunet cells (Figure 1A). Moreover, we also blotted the cell lysates from Huh7-Con1 (Con1) and Lunet cells with 8B4, an mAb specific for the N-terminus of PrP. We found that PrP was detectable in Huh7-Con1 cells at very low levels but could not be detected in Lunet cells (Figure 1B). To confirm that PrP was expressed at a higher level in Huh7-Con1 cells than in Lunet cells, we stained these cells with another PrP-specific mAb, 4H2, which is specific for the C-terminus of PrP and is highly sensitive for immunofluorescence staining applications. Indeed, we found that PrP expression was higher in Huh7-Con1 cells (Con1) than in Lunet cells (Figure 1C). Interestingly, some PrP was detected in the cytosol (Figure 1C). Taken together, these results suggested that HCV replication may upregulate the expression of PrP.
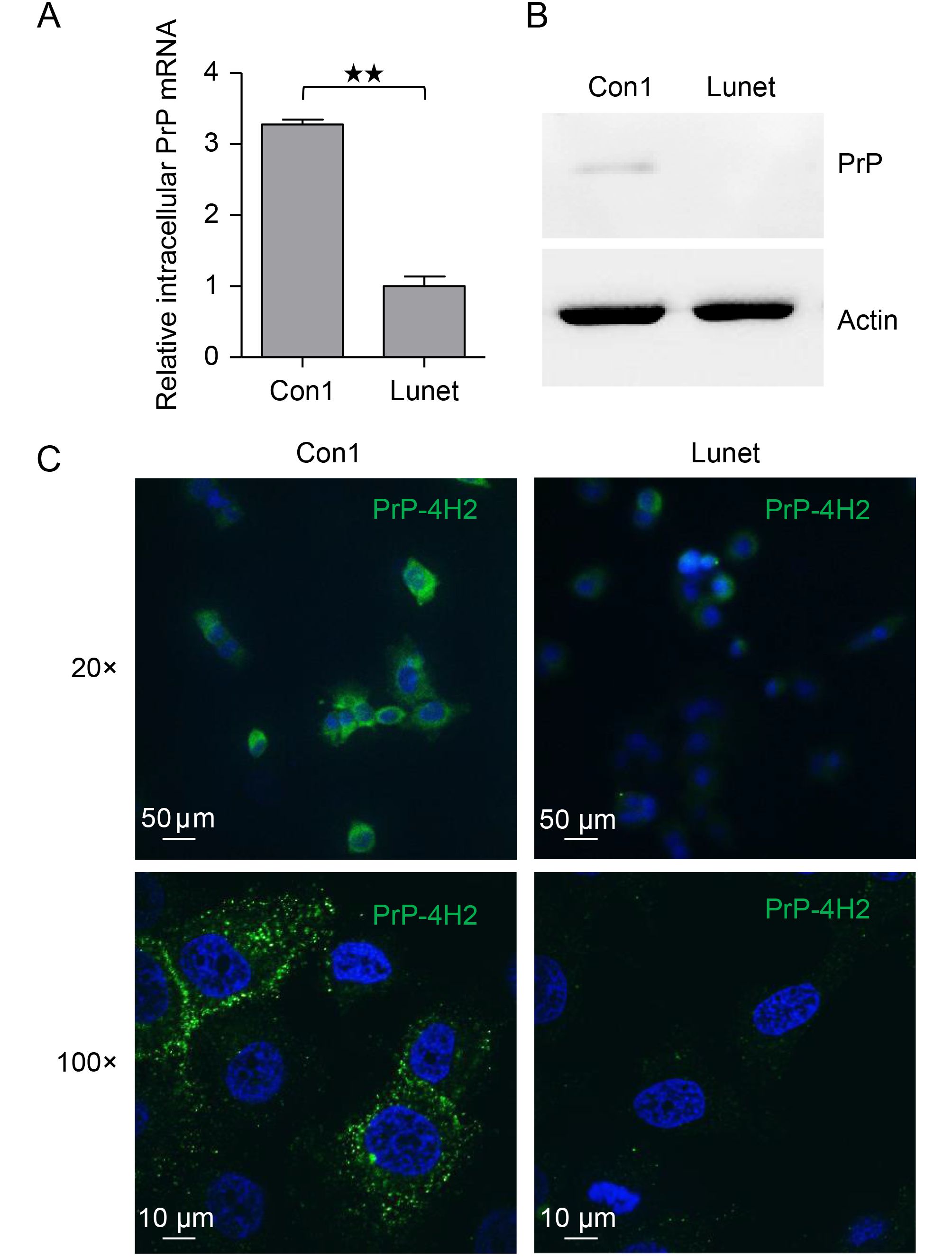
Figure 1. PrP expression was upregulated in Huh7-Con1 cells. (A) Intracellular PrP mRNA levels were quantified by qPCR in Huh7-Con1 (Con1) and Lunet cells. (B) Huh7-Con1 (Con1) and Lunet cell lysates were blotted with anti-PrP Abs (8B4). (C) Huh7-Con1 (Con1) and Lunet cells were stained with 4H2 mAbs, and bound Abs were detected with Alex Fluor488-conjugated goat anti-mouse Abs (green). Nuclei were counter-stained with DAPI (blue). Images were acquired using a 20× (upper panel) or 100× (bottom panel) optical lens. The experiments were repeated three times with similar results. Data are means ± SEMs. Two-tailed Student’s t-tests were performed for statistical analysis. **P < 0.01, *P < 0.05.
-
Next, we investigated the roles of PrP in HCV replication by overexpressing PrP in Huh7-Con1 cells. We detected an increase in PrP mRNA expression of more than 200-fold in Huh7-Con1 cells transfected with PrP (Con1-PrP) compared with that in Huh7-Con1 cells transfected with pHAGE empty vector (Con1-NC; Figure 2A). Next, we detected intracellular HCV RNA levels by qPCR. We found that HCV RNA was indeed significantly upregulated (Figure 2B) in PrP overexpressing Huh7-Con1 cells (Con1-PrP) compared with control Con1 cells (Con1-NC). To confirm that HCV replication was upregulated, we blotted cell lysates with Abs specific for NS3 and NS5B and found significant elevation in NS3 and NS5B levels in Con1-PrP cells (Figure 2C). We also confirmed significant PrP expression in Con1-PrP cells (Figure 2C). To determine whether the PrP overexpressed in Huh7-Con1 cells was GPI-anchored, we performed PI-PLC treatment in Con1-PrP cells. We found that most of the PrP overexpressed on the cell surface of Huh7-Con1 cells was resistant to PI-PLC treatment. As expected, the PrP expressed on the cell surface of WVT human neuroblastoma cells was sensitive to PI-PLC treatment (Figure 2D). These results suggested that overexpressed PrP in Huh7-Con1 cells may be pro-PrP.
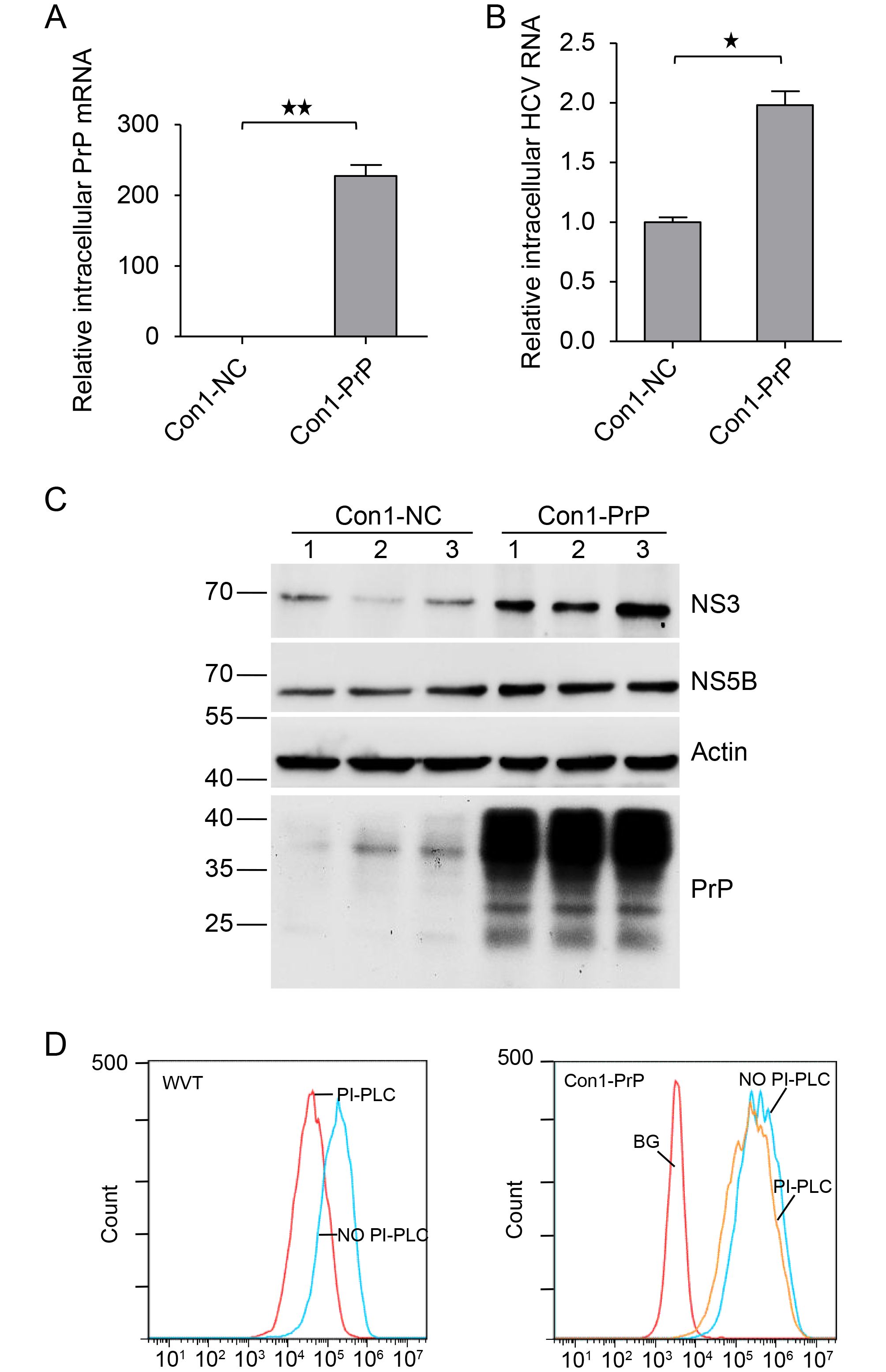
Figure 2. Overexpression of PrP upregulated HCV replication in Huh7-Con1 cells. Huh7-Con1 cells were transfected with pHAGE-PrP (Con1-PrP) or pHAGE empty vector (Con1-NC) and cultured for 48 h. (A and B) Total intracellular RNAs were extracted and subjected to qPCR to detect PrP mRNA (A) and HCV RNA (B) levels. (C) Total cell lysates were extracted and immunoblotted for PrP using anti-PrP specific mAbs (4H2), HCV NS3, and NS5B. Lanes 1–3 are representative of triplicate Con1-NC and Con1-PrP samples. (D) Con1-PrP and WVT cells were subjected to PI-PLC treatment and then analyzed using 8H4 mAbs or mouse IgG1 isotype control by flow cytometry. The experiments were repeated three times with similar results. Data are means ± SEMs. Two-tailed Student’s t-tests were applied for statistical analysis. **P < 0.01, * P < 0.05.
-
NS3 is a multifunctional protein, and its helicase domain is involved in HCV replication (Ma et al., 2008). NS5B is an RNA-dependent RNA polymerase (RdRp) and is also required for HCV replication (Yamashita et al., 1998; Simister et al., 2009). Upregulation of these two proteins in PrP-overexpressing Huh7-Con1 cells suggested that PrP may be involved in HCV replication. To gain further support for this interpretation, we performed confocal immunostaining of NS3/NS5B with PrP. We found that PrP and NS3 or PrP and NS5B were colocalized inside the cytosol with anti-PrP mAbs but not with an isotype control mAb (Figure 3). Because NS3 and NS5B are important proteins for HCV replication, these results implied that PrP was directly involved in the genomic replication of HCV.
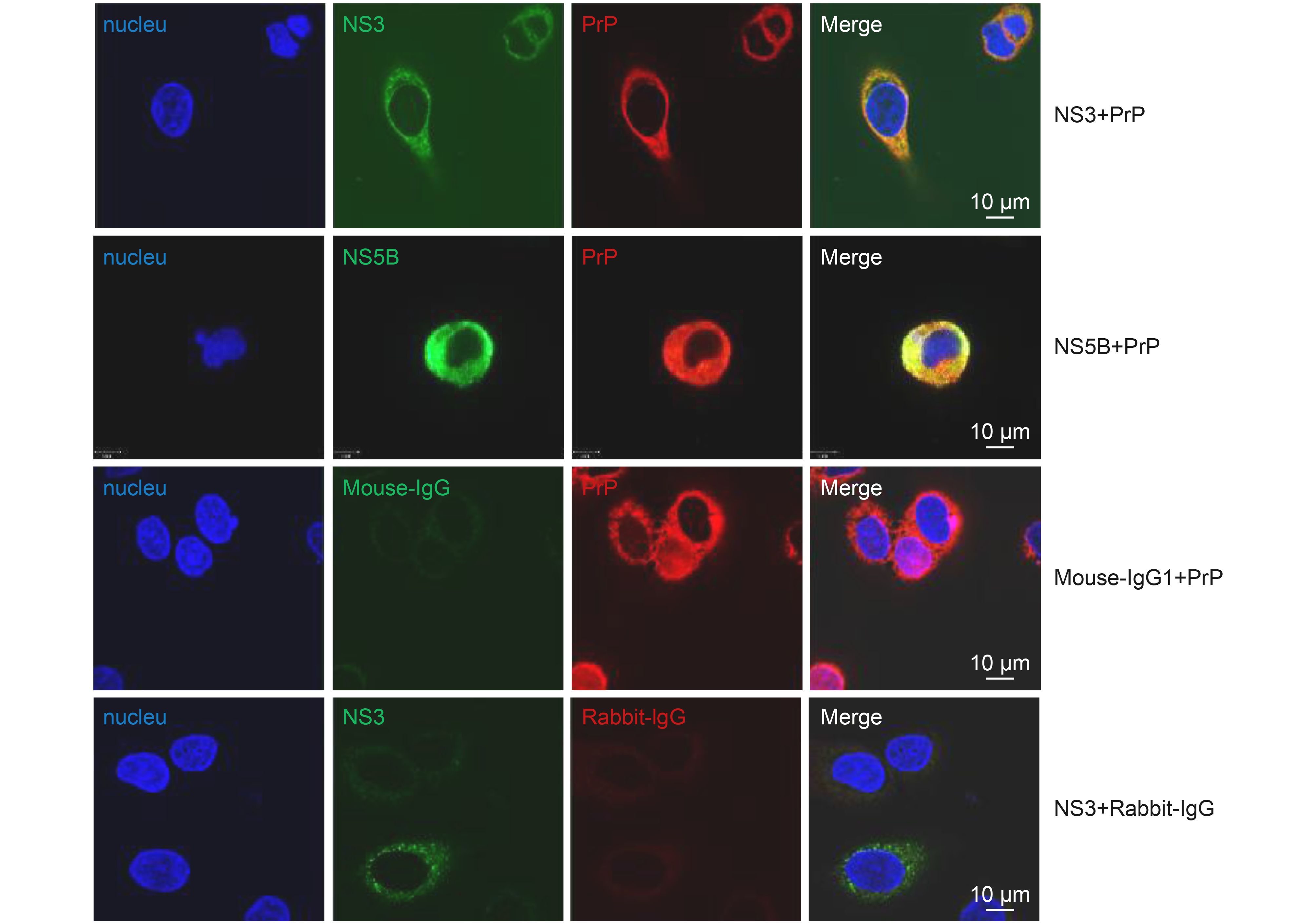
Figure 3. Colocalization of PrP with HCV NS3 and NS5B. Huh7-Con1 cells were transfected with WT PrP and then co-stained with PrP (red) and NS3 (green) or PrP and NS5B (green). Mouse IgG1 and rabbit IgG served as antibody isotype controls. Bound Abs were further detected with Alex Fluor-conjugated secondary Abs. Nuclei were counter stained with DAPI (blue).
-
PrP binds RNA and has DNA strand transfer properties (Gabus et al., 2001a; Gabus et al., 2001b). Alais et al. reported that the KKRPK motif of PrP can bind genomic RNA of HIV, thus reducing its translation (Alais et al., 2012). To investigate which motif of PrP is involved in enhancing HCV replication, we generated three PrP mutants (Figure 4A). We deleted the RNA-binding sequence KKRPK (Δ23–27) at the N-terminus of PrP and also deleted aa 97–102 (Δ97–102), which was close to the hydrophobic motif of PrP. This motif was shown to be able to bind the cell membrane. Finally, we deleted aa 226–231 (Δ226–231), which was contiguous to the GPI-PSS. Deletion of this motif results in loss of the GPI anchor. We then transfected these plasmids individually into Huh7-Con1 cells (Δ23–27, Δ97–102, and Δ226–231). The expression of PrP was confirmed by immunoblotting with 4H2 (Figure 4B). In fact, WT PrP was expressed at a relatively lower level than PrPΔ23–27 and PrPΔ97–102, and all constructs were expressed as two glycosylated forms and an unglycosylated form. In contrast, PrPΔ226–231 only showed one glycosylated form. Immunoblotting for NS3 and NS5B showed that in contrast to WT PrP, PrPΔ97–102, and PrPΔ226–231, PrPΔ23–27 did not support the enhanced HCV replication (Figure 4B). Since the aa 23–27 motif was composed of KKRPK, which can bind RNA and possesses RNA chaperon activity, these results suggested that loss of KKRPK led to the loss of elevated HCV RNA replication. Because PrP is colocalized with NS5B, the RdRp of HCV, and NS3, PrP may harness its KKRPK motif to bind HCV RNA and facilitate HCV replication.
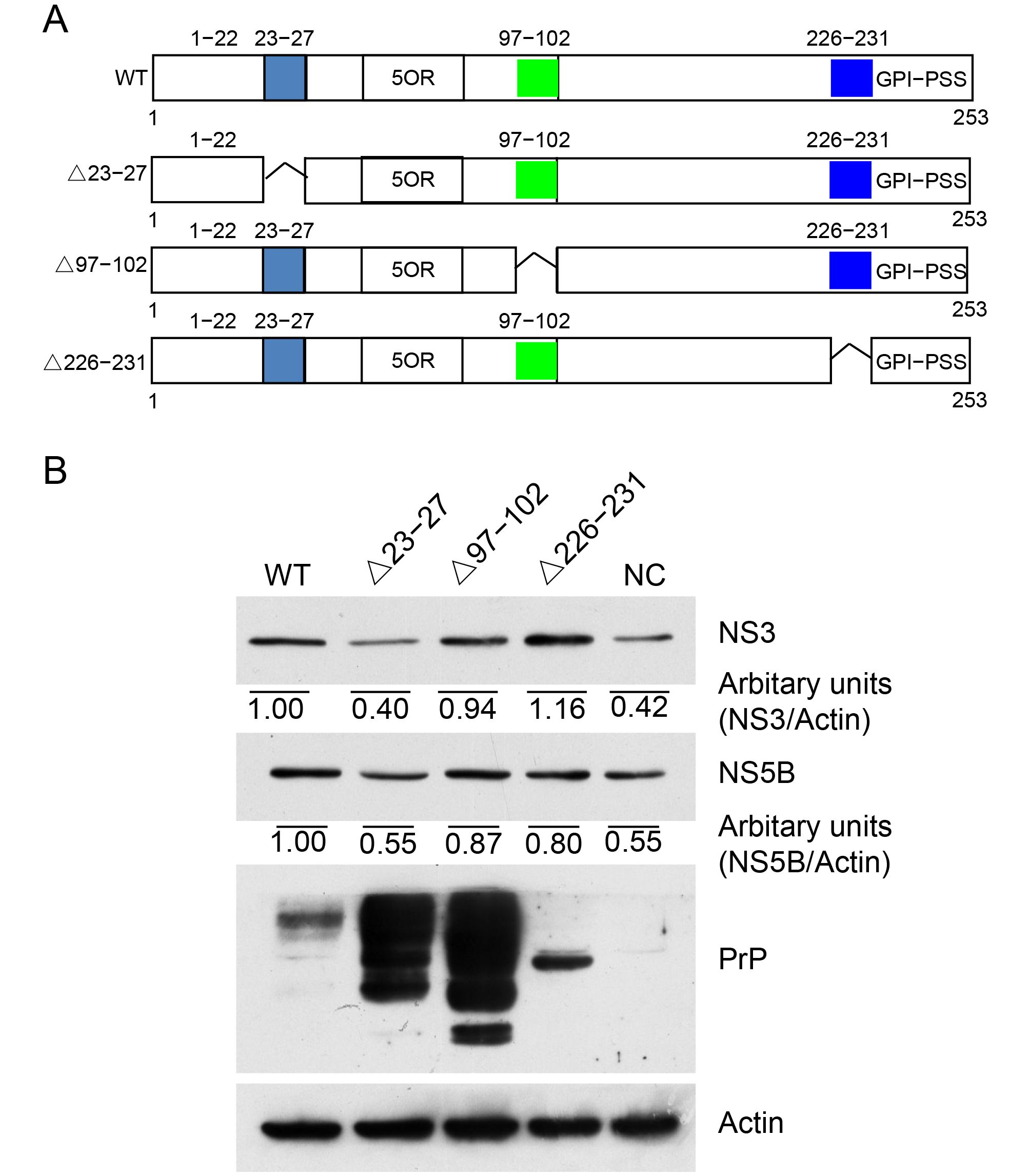
Figure 4. Amino acids 23–27 were essential for PrP-enhanced HCV replication. (A) A schematic diagram of WT and truncated PrP (Δ23-27 lacking aa 23–27; Δ97-102 lacking aa 97–102; Δ226-231 lacking aa 226–231). (B) Huh7-Con1 cells were transfected with WT PrP (WT), corresponding truncated PrP constructs (Δ23-27, Δ97-102, and Δ226-231), or empty vector (NC) and cultured for 48 h. Total cell lysates were extracted and immunoblotted for PrP, HCV NS3, and NS5B.
-
Chronic HCV infection usually leads to hepatosteatosis, fibrosis, cirrhosis, and eventually hepatocellular carcinoma. The successful development of anti-HCV drugs, such as sofosbuvir/grazoprevir, has revolutionized HCV treatment and was hailed as “the end of the game” for HCV research (Manns and Cornberg, 2013; Kwo et al., 2017). However, in addition to being quite expensive, recent studies have suggested that curing HCV may re-activate hepatitis B virus (HBV) in patients infected with HCV and HBV (Collins et al., 2015; Ende et al., 2015; Odolini et al., 2017). Thus, further studies of HCV biology are needed to identify novel drug targets.
In this study, we found that HCV replication enhanced PrP expression to further promote HCV replication. This conclusion was based on the following observations: (1) PrP expression was detected in Huh7-Con1 cells but not Lunet cells; (2) overexpression of PrP in Huh7-Con1 cells resulted in higher levels of HCV RNA and proteins; (3) PrP colocalized with NS5B, the RdRp of HCV; and (4) deletion of the RNA/DNA binding motif KKRPK on PrP abolished the function of PrP to enhance HCV expression. However, at the present time, the underlying mechanisms through which HCV replication enhances PrP transcription are still unclear. Moreover, it is unknown whether our observations in the Con1 replicon system may be applicable to other cell models or in animals.
HCV replication complexes are normally associated with the cytoskeleton (Lai et al., 2008), which is composed of both actin and tubulin. In Huh7-Con1 cells, expressed PrP was most likely present as pro-PrP, which binds to FLNa via its GPI-PSS. Although we did not prove that PrP indeed was pro-PrP in this case, two observations supported our assumption. First, some PrP in Huh7-Con1 cells could be detected in the cytosol, suggestive of pro-PrP. In addition, cell surface PrP was resistant to PI-PLC treatment, suggesting that PrP may not have a GPI anchor. Thus, we proposed a working model to describe how PrP expression enhances HCV replication. Inside the cytoplasm, pro-PrP uses its GPI-PSS to bind FLNa, which crosslinks actin. This binding provides anchoring points for PrP, which then binds HCV RNA with its KKRPK motif. This complex also recruits HCV NS3 and NS5B to facilitate HCV replication (Figure 5).
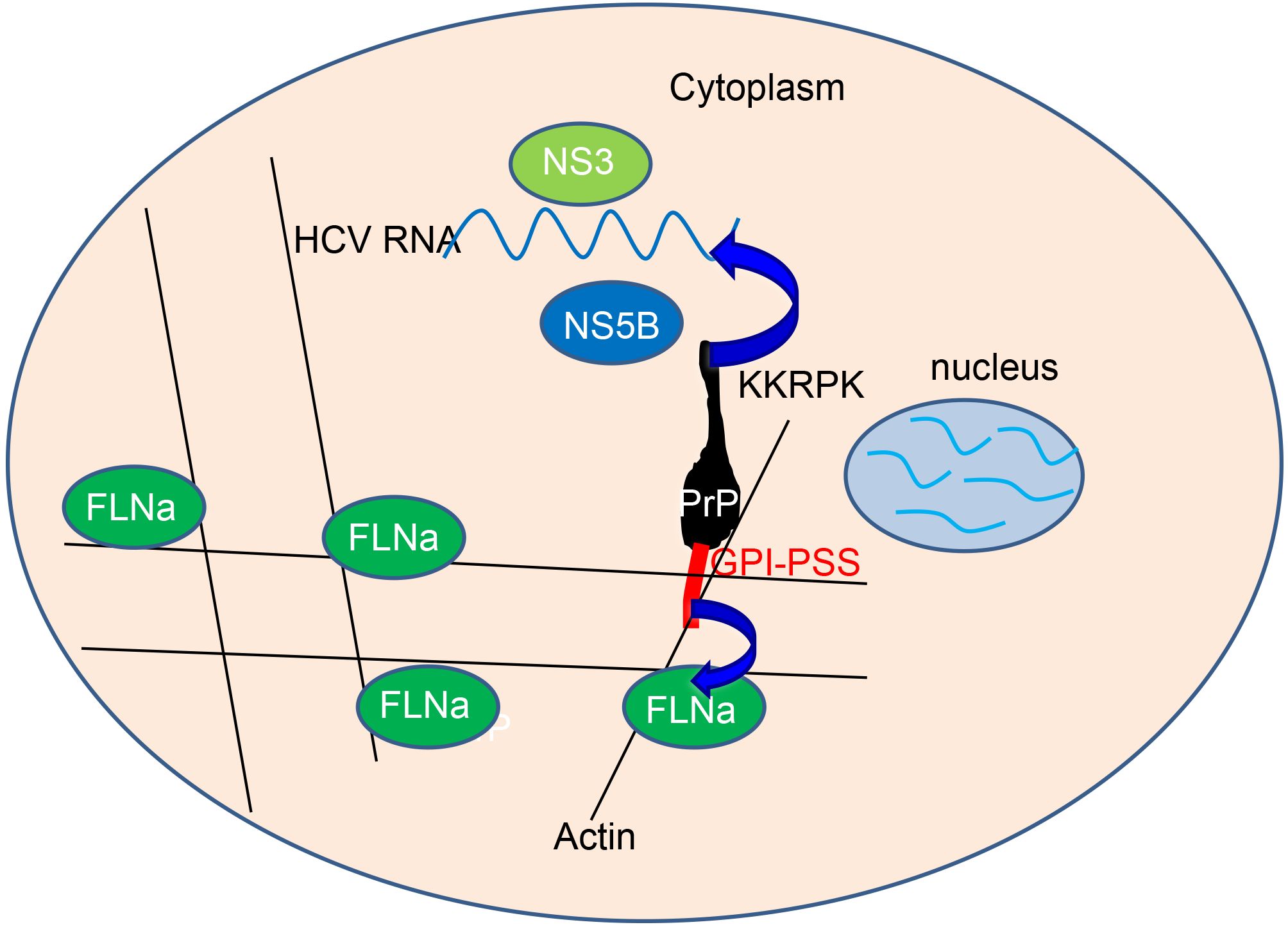
Figure 5. Model describing how PrP expression enhanced HCV replication. Inside the cytoplasm, pro-PrP uses its GPI-PSS to bind FLNa, which crosslinks with the actin cytoskeleton. This binding provides anchoring points for PrP, which then binds HCV RNA with its KKRPK motif. This complex also recruits NS3 and NS5B to facilitate HCV replication.
Although PrP is widely expressed in many tissues, no PrP is detected in normal hepatocytes. In contrast, PrP is one of the earliest expressed proteins in activated stellate cells following liver injury (Ikeda et al., 1998). In addition, some HBV-induced hepatocellular carcinoma cells were found to be positive for PrP (data not shown). These observations suggested that PrP may be induced in hepatocytes infected by HBV. PrP expression has also been observed in other tissues infected with different viruses (Nikles et al., 2005; Lötscher et al., 2007; Caruso et al., 2009). However, whether HCV infection activates the expression of PrP is still unclear. Based on the observation that PrP expression was upregulated in Huh7-Con1 cells compared with Lunet cells, it is likely that HCV infection may stimulate the expression of PrP in hepatocytes.
In contrast to previous reports demonstrating that PrP expression inhibits virus replication, we found that PrP expression was associated with enhanced HCV RNA replication, as supported by the observation that PrP colocalized with NS5B, the HCV RdRp for RNA replication. However, at the present time, it is unknown whether PrP affects HCV replication in the context of infection. Thus, further studies are needed to determine the specific role of PrP in HCV infection.
These findings are potentially important for understanding the interactions between host factors and HCV replication, and may provide a potential therapeutic target for HCV infection.
-
This work was supported by the Strategic Priority Research Program A of the Chinese Academy of Sciences (XDA12010309), the National Science Foundation of China (31670170), the Nature Science Foundation of Hubei Province (2015CFA087), and the National Basic Research Priorities Program of China (2013CB911102). We thank the Core Facility and Technical Support, Wuhan Institute of Virology for assistance with confocal microscopy (Dr. Ding Gao) and flow cytometry (Ms. Juan Min).
-
The authors declare no competing financial interests. This article does not contain any studies with human or animal subjects.
-
CYL and HXZ designed the study, HXZ performed experiments. CYL, HXZ, SSG, RJP, XWC, analyzed the data. CYL and HXZ wrote the paper.
Supplementary Table S1 is available on the websites of Virologica Sinica: www.virosin.org; link.springer.com/journal/12250.
-
Primer Sequence PrPΔ23-27/F 5′-AGTGACCTGG GCCTCTGCCC TGGAGGATGG AACACTG-3′ PrPΔ23-27/R 5′-CAGTGTTCCA TCCTCCAGGG CAGAGGCCCA GGTCACT-3′ PrPΔ97-102/F 5′-AGGAGGTGGCACCCACAGTAAGCCAAAAACCA-3′ PrPΔ97-102/R 5′-TGGTTTTTGGCTTACTGTGGGTGCCACCTCCT-3′ PrPΔ226-231/F 5′-GGGAATCTCAGGCCTATATGGTCCTCTTCTCC-3′ PrPΔ226-231/R 5′-GGAGAAGAGGACCATATAGGCCTGAGATTCCC-3′ q HCV (Con1)/F 5′-ATCACTCCCCTGTGAGGAACT-3′ q HCV (Con1)/R 5′-GCGGGTTGATCCAAGAAAGG-3′ q PrP/F 5′-GACCTGGGCCTCTGCAAGAAG-3′ q PrP/R 5′-TGAGGTGGGTAGCGGTTGCC-3′ q GAPDH/F 5′-GAAGGTGAAGGTCGGAGTC-3′ q GAPDH/R 5′-GAAGATGGTGATGGGATTTC-3′ Table S1. Primers used for amplification and qPCR
Hepatitis C virus-induced prion protein expression facilitates hepatitis C virus replication
- Huixia Zhang 1,2 ,
- Shanshan Gao 1,2 ,
- Rongjuan Pei 1 ,
- Xinwen Chen 1 ,
-
Chaoyang Li
1,,

- Received Date: 22 June 2017
- Accepted Date: 18 September 2017
- Published Date: 25 October 2017
Abstract: Hepatitis C virus (HCV) infects approximately 180 million people worldwide. Significant progress has been made since the establishment of in vitro HCV infection models in cells. However, the replication of HCV is complex and not completely understood. Here, we found that the expression of host prion protein (PrP) was induced in an HCV replication cell model. We then showed that increased PrP expression facilitated HCV genomic replication. Finally, we demonstrated that the KKRPK motif on the N-terminus of PrP bound nucleic acids and facilitated HCV genomic replication. Our results provided important insights into how viruses may harness cellular protein to achieve propagation.

 DownLoad:
DownLoad: