










-
Dear Editor,
Mammalian cells encode five members of the nucleosome assembly protein 1 (NAP1) family of proteins including nucleosome assembly protein 1-like 1 (NAP1L1), NAP1L2, NAP1L3, NAP1L4 and NAP1L5 (Attia et al. 2011). NAP1L1 is ubiquitously expressed and has been implicated in several processes of transcription and DNA replication, such as the nucleosome assembly, nucleosome sliding, transcriptional activation through chromatin remodeling, and nucleocytoplasmic shuttling of histones (Lee et al. 2017; Moshkin et al. 2009; Zlatanova et al. 2007). Furthermore, NAP1L1 is involved in the life cycle of several DNA viruses (EBV, KSHV and HPV) (Gupta et al. 2016; Rehtanz et al. 2004; Wang and Frappier 2009) and retroviruses (HIV and HTLV) (Sharma and Nyborg 2008; Vardabasso et al. 2008). An interesting but yet unanswered question is whether NAP1L1 is involved in the life cycle of RNA viruses.
Hepatitis C virus (HCV) encoded a 9.6-kb positivesense RNA genome and is classified as the Hepacivirus genus of the Flaviviridae family. HCV affects 71 million people worldwide, which is a major human pathogen that causes chronic liver disease. HCV polyprotein is processed into 10 proteins in the following order: core, E1, E2, p7, NS2, NS3, NS4A, NS4B, NS5A, and NS5B (Moradpour et al. 2007).
HCV NS3 protein consists of a proteinase domain NS3(1–180) and a helicase domain NS3(181–631). To determine the cellular interacting partners of HCV NS3 and gain insight into its cellular function, GST pulldown coupled to mass spectrometry was used. Constructs encoding for NS3(1–180) from JFH1 (genotype 2a strain of HCV) were appended to the carboxyl terminus of glutathione-s-transferase (GST) and were generated using pGEX4T-1 expression plasmids (Amersham Biosciences, Piscataway, NJ, USA). Plasmid transfected into Huh7.5.1 cells was performed using FuGENE HD (Promega, Madison, WI, USA) according to the manufacturer instructions. The target protein NS3(1–180) was coupled to GST to generate a fusion protein in bacterial. The fusion protein was accumulated onto glutathione beads and incubated with Huh7.5.1 cell lysates for pulldown assay. For mass spectrometry identification, protein complexes pulled down from Huh7.5.1 cells were sent to Beijing Protein Innovation. Potential interacting partners for NS3(1–180) was identified by mass spectrometry. Interestingly, NAP1L1 was in the list of NS3(1–180) binding partners.
To confirm the interaction between NS3 and NAP1L1, lysates from Huh7.5.1 cells were incubated with GSTNS3(1–180), or GST-NS3(181–631). Bound proteins were detected by immunoblotting using anti-NAP1L1 antibody (Abcam, Cambridge, MA, USA, Catalogue no. ab33076). The result showed that NS3(1–180) but not NS3(181–631) was able to pull down NAP1L1 (Fig. 1A). To further validate the interaction between NS3 and NAP1L1, 293T cells were transiently transfected with constructs expressing FLAG-tagged HCV proteins from JFH1 or the empty expression vector. Immuno-precipitation experiments using whole cell lysates were performed as previously described (Li et al. 2014; Yin et al. 2016). As shown in Fig. 1B, the FLAG antibody (Sigma-Aldrich, Piscataway, NJ, USA, Catalogue no. A2220) was able to precipitate NAP1L1 from extracts of cells transfected with the vector for FLAG-tagged NS3 and not from extracts of cells transfected with the empty expression vector or with a vector expressing other HCV proteins including NS4A and NS5A. Recent studies described the interaction of HCV NS5A and NAP1L1 (Cevik et al. 2017; Goonawardane et al. 2017). However, NS5A from genotype 2a didn't associated with NAP1L1 in our study. We hypothesized that NAP1L1 might associate with NS5A in a genotypedependent manner. 293T cells were transfected with V5-NS5A-1a, V5-NS5A-1b, V5-NS5A-2a, V5-NS5A-3a, V5-NS5A-3b, or empty vector for 24 h. Cell lysates were incubated with anti-V5 antibody-coated beads (Invitrogen, Carlsbad, CA, USA, Catalogue no. 14-6796-82). Interestingly, we found NS5A from genotype 1a, 1b, 3a and 3b interacted with NAP1L1 (Fig. 1C). Thus, the interaction between NS5A and NAP1L1 is genotype dependent.
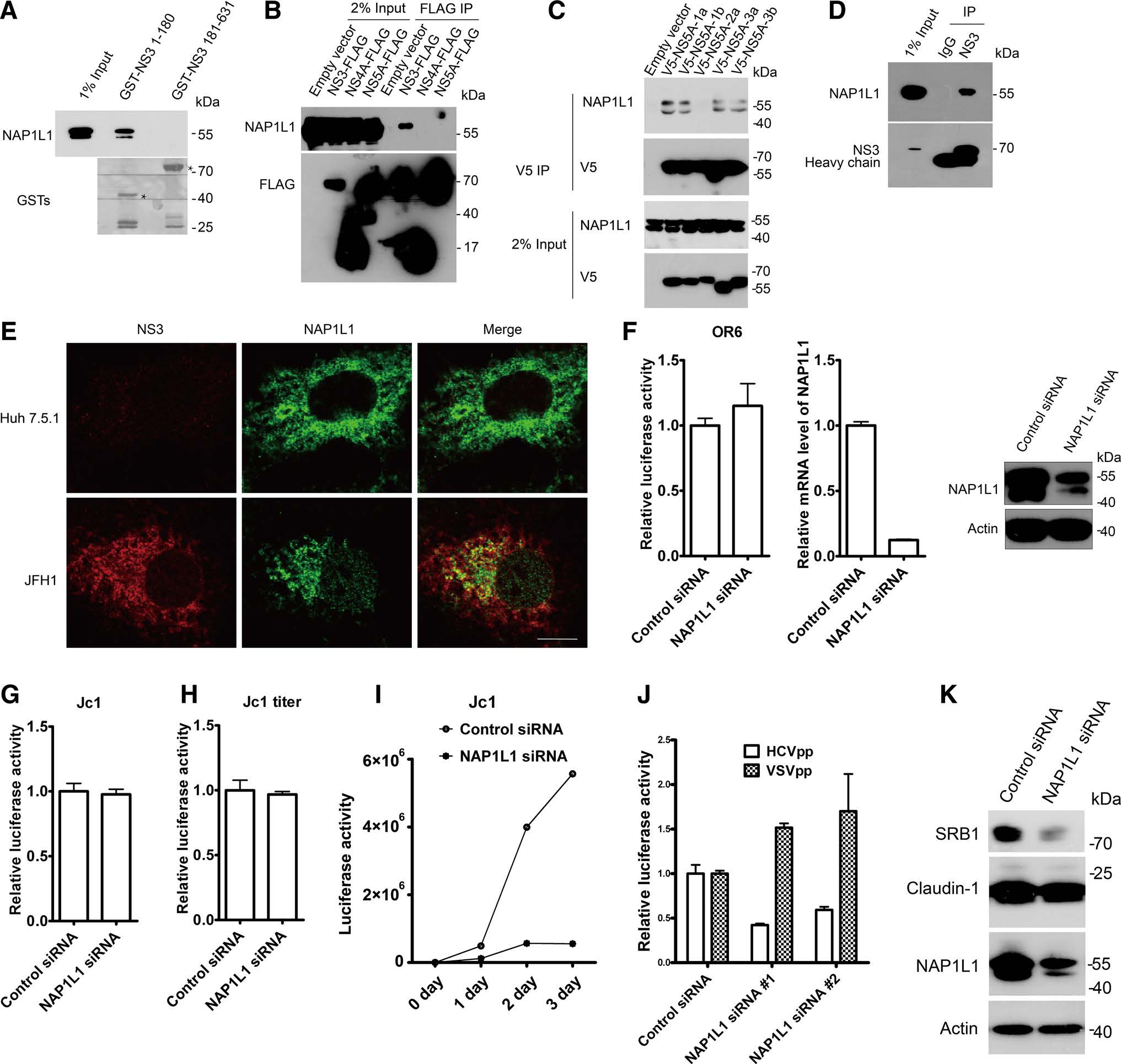
Figure 1. NAP1L1 associates with HCV NS3 and regulates HCV entry. A Cell lysates from Huh7.5.1 cells were incubated with purified GSTNS3(1–180) or GST-NS3(181–631) and proteins were pulled down with glutathione-Sepharose beads. Bound proteins were detected by immunoblotting using anti-NAP1L1 antibody. GST fusion proteins were marked by an asterisk. B 293T cells were transfected with NS3-FLAG, NS4A-FLAG, NS5A-FLAG or empty vector for 24 h. Cell lysates were incubated with anti-FLAG antibody-coated beads and co-IP proteins were subjected to Western blotting using anti-NAP1L1 antibody and anti-FLAG antibody. C 293T cells were transfected with V5-NS5A-1a, V5-NS5A-1b, V5-NS5A-2a, V5-NS5A-3a, V5-NS5A-3b, or empty vector for 24 h. Cell lysates were incubated with anti-V5 antibody-coated beads and co-IP proteins were subjected to Western blotting for analysis. D Cell lysates from Jc1 infected Huh7.5.1 were immunoprecipitated with anti-NS3 antibody or control IgG followed by Western blot. E Huh7.5.1 cells infected with JFH1 for 72 h were stained with indicated antibodies. Scale bar: 10 lm. F OR6 cells were transfected with siRNA targeting NAP1L1 for 72 h and then Renilla luciferase activity and cellular ATP levels were measured. Knockdown efficiency of NAP1L1 was validated by quantitative PCR and Western blot with anti-NAP1L1 antibody and anti-actin antibody. F, G Huh7.5.1 cells were infected with Jc1 for 3 days and then treated with siRNA against NAP1L1 or control siRNA for 3 days. The infection activity and ATP levels were measured. The relative luciferase activity was normalized to the cellular ATP levels (G). The supernatants were tittered in H. I Huh7.5.1 cells were treated with siRNA against NAP1L1 or control siRNA for 3 days and then infected with Jc1. The infection activity of different time was measured. J Huh7.5.1 cells transfected with siRNAs against NAP1L1, or with control siRNA for 3 days were infected with genotype 1b HCVpp (Con1) or VSVpp for another 3 days, followed by measurement of the firefly luciferase activity and cellular ATP levels. The relative luciferase activity was normalized to the cellular ATP level. Data are presented as the mean ± SD. K Cell lysates from Huh7.5.1 cells treated with NAP1L1 siRNA #1 or control siRNA for 3 days were immunoblotted for indicated antibodies.
To investigate the interaction between NS3 and NAP1L1 during HCV infection, we infected Huh7.5.1 cells with Jc1GLAG2 (p7-nsGluc2A), a genotype 2a strain of infectious HCV (from Dr. Charles Rice). Lysates from infected Huh7.5.1 cells were immunoprecipated with NS3 antibody (Abcam, Cambridge, MA, USA, Catalogue no. ab65407) or control IgG (MBL, Nagoya, Japan, Catalogue no. M075-3). As shown in Fig. 1D, NS3 antibody was able to precipitate endogenous NAP1L1, confirming the interaction between NS3 and NAP1L1.
The specific interaction of HCV NS3 with NAP1L1 prompted us to examine whether these proteins colocalized within infected cells by immunofluorescence microscopy. Huh7.5.1 cells infected with JFH1 (from Dr. Takaji Wakita) were subjected to immunofluorescence staining using rabbit anti-NAP1L1 and mouse anti-NS3 antibody. Immunofluorescence microscopy experiments were performed as previously described (Zhang et al. 2016). As shown in Fig. 1E, in JFH1-infected Huh7.5.1 cells, NAP1L1 partially colocalized with HCV NS3 protein.
To investigate the role of NAP1L1 in HCV life cycle, we applied siRNA strategy to silence NAP1L1. To detect the mRNA transcription of NAP1L1, quantitative PCR (qPCR) was performed as previously described (Li et al. 2014). Sequences of primers used in qPCR were as follows: The forward and reverse primers for GAPDH were 50-ACCTTCCCCATGGTGTCTGA-30 and 50-GCTCCTC CTGTTCGACAGTCA-30; the forward and reverse primers for NAP1L1 were 50-AAAGCACGTCAGCTAACTGT-30 and 50-TTGAGAGCATTCACTCGTCTTTT-30. The OR6 replicon cell line, obtained from Drs. Nobuyuki Kato and Masanori Ikeda, is a successful model of HCV replication, which contains a full-length genotype 1b HCV RNA with the Renilla luciferase as a reporter. The Renilla luciferase activity reflected the amount of HCV RNA synthesized, and the cell viability was evaluated by assessing cellular ATP levels. To determine the effect of NAP1L1 for HCV replication, OR6 cells were seeded in 96-well plates for 24 h and then treated with siRNA against NAP1L1 (NAP1L1 siRNA #1: AAGGAACACGAUGAACCUAUU) for another 72 h. Knockdown efficiency of NAP1L1 was validated by quantitative PCR and Western blot with antiNAP1L1 antibody and anti-actin antibody (Sigma-Aldrich, Piscataway, NJ, USA, Catalogue no. A2228). In OR6 replicon cells, silencing NAP1L1 had no effect on HCV replication, indicating that NAP1L1 is not required for HCV replication (Fig. 1F).
In Jc1GLAG2(p7-nsGluc2A)-infected Huh7.5.1 cells, HCV replication measured by Gaussia luciferase reporter and the titer of HCV in the supernatant remained the same when we treated the cells with NAP1L1 siRNA after HCV infection (Fig. 1G, H). However, HCV replication was reduced when NAP1L1 is knocked down by siRNA 3 days before HCV infection (Fig. 1I). Thus, we hypothesized that NAP1L1 is important for HCV entry. To address that, we silenced NAP1L1 by two NAP1L1 siRNA (NAP1L1 siRNA #1: AAGGAACACGAUGAACCUAUU, NAP1L1 siRNA #2: GGUAGAAACACCAACAGGAUACAUU) and examined its effect on HCV pseudoparticles (HCVpp) entry. HCVpp or VSV pseudovirus particles (VSVpp) were generated in 293T cells by transfection with constructs expressing HIV gag/pol (pLP1), HIV rev (pLP2), pLenti6 encoding luciferase, and HCV E1E2 from genotype 1b strain Con1 or the VSV-G expression plasmid (gifts from Dr. Ping Zhao). Knocking down NAP1L1 by siRNA inhibited the entry of HCVpp but not VSVpp (Fig. 1J). Taken together, our results indicate that NAP1L1 is critical for HCV entry.
Next, we examined whether protein levels of HCV coreceptors were changed in NAP1L1 depleted cells. As shown in Fig. 1K, protein level of SR-B1 but not claudin-1 was reduced by NAP1L1 siRNA #1, which partially explained how NAP1L1 was involved in HCV entry.
In our study, we used siRNA against NAP1L1 to dissect the role of NAP1L1 in HCV life cycle. Knocking down NAP1L1 will reduce de novo HCV infection, but not established HCV infection. Those interesting results led us to identify the key role of NAP1L1 in HCV entry. During the preparation stage of our manuscript, two papers on the role of NAP1L1 in HCV came out (Cevik et al. 2017; Goonawardane et al. 2017). One paper suggested that NAP1L1 is required for HCV replication (Goonawardane et al. 2017), while the other paper demonstrated that NAP1L1 does not participated in HCV replication (Cevik et al. 2017). In consistent with the second paper (Cevik et al. 2017), we found that NAP1L1 is not required for HCV replication, but important for HCV entry.
Through GST pulldown coupled with mass spectrometry, we identified NAP1L1 as a novel binding partner for HCV NS3. NS3 is not present in the cells before HCV has entered the cells. Although NAP1L1 depletion acts on HCV entry, this cannot be due to the interaction with NS3 but must be another effect. In future, more work need to be done to explore how NAP1L1 is involved in HCV entry and to dissect the functional role of NS3-NAP1L1 interaction.
In summary, we identified NAP1L1 as a novel binding partner of HCV NS3 and found that the protease domain of NS3 is responsible for interactions with NAP1L1. We demonstrated the functional role of NAP1L1 in HCV entry through regulating the protein expression of SR-B1. Once HCV replication is established, NAP1L1 is no longer required for viral replication. Our studies provide novel insights into the role of NAP1L1 in RNA virus life cycle.
HTML
-
This work was supported by grants from the Beijing Natural Science Foundation (7164283), National Natural Science Foundation of China (81471955, 81672035), PUMC youth Fund (3332016085), the Fundamental Research Funds for the Central Universities, CAMS Innovation Fund for Medical Sciences (CIFMS) (2016-I2M-3-020), and Program for Changjiang Scholars and Innovative Research Team in University (IRT13007). We thank Charles Rice, Francis Chisari, Ping Zhao, Masanori Ikeda, Nobuyuki Kato and Takaji Wakita for reagents.
-
The authors declare that there are no conflicts of interest.
-
This article does not contain any studies with human or animal subjects performed by any of the authors.

 DownLoad:
DownLoad: