












HTML
-
Coxsackievirus group B (CVB) belongs to the genus Enterovirus of the Picornaviridae family, which is one of the main pathogens of viral myocarditis and dilated cardiomyopathy (Kemball et al. 2009). At present, the exact mechanism of CVB-induced heart disease remains unclear (Luo et al. 2002; Rassmann et al. 2006). Clinically, there are few effective medications and measures to prevent and treat myocardial diseases caused by CVB type 3 (CVB3) infection. Studies have shown that aspects such as the shutdown of host cellular protein synthesis, enhanced permeability of the plasma membrane, and dysregulated singling pathways all contribute the pathogenicity of CVB3 (Kemball et al. 2009; Luo et al. 2002; Rassmann et al. 2006; Chau et al. 2007); however, the pathogenesis of viral myocarditis cannot be explained by these outcomes alone.
In recent years, studies have been focusing on the interactions between virus and the host, in particular, the role of stress granules (SGs) in viral infections (Lindquist et al. 2010; Piotrowska et al. 2010). The transcription, translation, localization, and degradation processes of viral genomes may all closely related to SG formation (Miller 2011; Panas et al. 2012; White and Lloyd 2012; Lloyd et al. 2013). SGs are granular aggregations formed in the cytoplasm when eukaryotic cells are under environmental stress, such as heat shock, oxidative stress (Palangi et al. 2017), ultraviolet radiation (Moutaoufik et al. 2014), and viral infection (Miller 2011). Viral infection can induce the formation of SGs, which in turn promote or inhibit viral infection (Piotrowska et al. 2010; Miller 2011). Some species of single-stranded, positive RNA viruses such as West Nile virus, Dengue virus, poliovirus, and hepatitis C virus were reported to induce the assembly of SGs (Pager et al. 2013; Hashimoto et al. 2016; Fernandez-Carrillo et al. 2018). However, the molecular mechanism underlying viral-induced SG formation remains unknown.
SGs are composed of untranslated mRNA, 40S ribosomal subunits, translation initiators (including eukaryotic translation initiation factor (eIF) 4E, eIF3, eIF2, eIF4A, eIF4G, and eIF4B), and RNA binding proteins such as poly(A)-binding protein), T cell intracellular antigen 1 (TIA-1), TIA1-related protein, human antigen R (HuR), fragile X mental retardation protein, tristetraprolin, and Ras GTPase-activating protein-binding protein 1 (G3BP1) (Kedersha et al. 2008; Kedersha and Anderson 2009). The dot-like aggregates of TIA1, HuR, or G3BP1 around the nucleus are often regarded as a marker of SG formation (Panas et al. 2012; Lloyd 2013; Hashimoto et al. 2016; Lindquist et al. 2011). During viral infection, the virus can regulate the formation of SGs to promote its own replication (Miller 2011). However, detailed mechanisms for viral-induced SG formation need to be further explored. In this study, the role of SGs in CVB3 infection was evaluated. Our data suggest that the biosynthesis of CVB3 is downregulated by the formation of SGs, especially at the early stage of viral infection.
-
HeLa cells were maintained in Dulbecco's modified Eagle's medium (DMEM) (Invitrogen, Carlsbad, CA, USA) supplemented with 8% (growth medium) or 5% (maintaining medium) fetal bovine serum (FBS) (Biological Industries, Beit Ha Emek, Israel) and antibiotics (50 U/mL penicillin and 50 mg/mL streptomycin) at 37 ℃ with 5% CO2. The cell line stably expressing EGFP-TIA1 was constructed and designated as HeLaEGFP-TIA1, which were grown in DMEM supplemented with 10% FBS and G418 (500 μg/mL). CVB3 Woodruff strain was passaged in HeLa cells and titrated using the plaque assay. EGFPCVB3, a CVB3 variant, was recovered by transfecting HeLa cells with pEGFP-CVB3.
-
First, a pcDNA3.1-based EGFP-expression vector was constructed as previously described (Wu et al. 2014a). Briefly, the EGFP coding sequence was obtained using polymerase chain reaction (PCR) from pEGFP-N1 (Clontech, Mountain View, CA, USA) using the sense primer and the antisense primer. Both pcDNA3.1 (Invitrogen) and the amplified EGFP fragment were digested using Nhe Ⅰ and Hind Ⅲ. The products of the digestion were linked and designated as pEGFP-C1. TIA1 and G3BP1 cDNA were also amplified by PCR, and plasmids were constructed by linking TIA1 and G3BP1 with pEGFP-C1. Similarly, a mCherry-HuR-expressing plasmid was constructed. All constructs were confirmed by restriction digestion and sequencing.
-
Cells were transfected using Lipofectamine 2000 (Invitrogen) according to the manufacturer's instructions. Briefly, HeLa cells were seeded in culture plates and grown to ~60% confluence. Cells were then transfected with siRNA or plasmid vectors and after 5 h in culture the medium was replaced with fresh medium.
-
HeLaEGFP-TIA1 cells were transfected with pmCherry-HuR for 24 h and then treated with sodium arsenite (Ars) (Sigma–Aldrich, Shanghai, China) at a final concentration of 0.5 mmol/L for 1 h and infected with CVB3 for various time periods (4, 5 h). The cells were washed three times with PBS and stained with Hoechst 33258 (0.4 g/mL in PBS). Images were viewed using a CV1000 confocal microscope (Yokogawa Electric, Tokyo, Japan) or an Axiovert 200 fluorescence microscope (Carl Zeiss, Gottingen, Germany). SG formation was measured by counting the number of cells that contained three or more granules.
-
The proteins from treated HeLa cells were extracted using Pierce RIPA buffer (Thermo Fisher, Shanghai, China) with PMSF cocktail (diluted at 1:100) (Abcam, Shanghai, China). Extracted proteins (20 μg) were analyzed using sodium dodecyl sulfate–polyacrylamide gel electrophoresis, transferred to a polyvinylidene fluoride membrane (0.45 μm; Millipore, Billerica, MA, USA), and incubated with primary antibodies overnight at 4 ℃. After a standard wash, the membrane was incubated with horseradish peroxidase labeled secondary antibody for 1 h at room temperature and washed again. The blots were stained using a SuperSignal kit (Pierce, Rockford, IL, USA) and imaged with a charge-coupled camera LAS4000 (Fujifilm, Tokyo, Japan). VP1 was detected with a monoclonal mouse anti-enterovirus (clone5-D8/1) VP1 antibody (Dako, Glostrup, Denmark) diluted at 1:1000. EGFP, eIF2α, and p-eIF2α antibodies (Cell Signaling, Beverly, MA, USA) were diluted at 1:1000. β-actin, used as a loading control, was detected using a polyclonal antibody (sc-130301, Santa Cruz, Shanghai, China).
-
HeLa cells were plated in 96-well plates at 2000 cells per well and were transfected with siRNA; control cells were transfected with scramble siRNA (si-Control). Briefly, 0.2 μL of Lipofectamine 2000 and 10 pmol of siRNA were added to each well. Cells were incubated for 5 h at 37 ℃ and culture medium was then replaced with medium containing 5% serum. Cells were then further cultured for another 48 h and the medium was replaced with fresh medium (180 μL/well). 3-(4, 5-dimethythiazol-2-yl)-2, 5-diphenyl tetrazolium bromide (MTT) solution (20 μL; 5 mg/mL in PBS) was added and the culture plate was incubated for 4 h at 37 ℃ in an incubator containing 5% CO2. After incubation, 150 μL of DMSOwas added to each well of thecultureplate, which was then placed on a shaker to thoroughly mix the solution. The absorbance of the culture plate was measured using ELISA reader at a wavelength of 490 nm.
-
Total RNA was extracted from HeLa cells using TRIzol reagent (Invitrogen) as described previously (Wu et al. 2014a, b). Total RNA (1 μg) was used as a template for RT along with antisense primers and PrimeScript RT Enzyme Mix Ⅰ (TaKaRa, Otsu, Shiga, Japan). PCR was performed with 1 μL of the synthesized cDNA, SYBR PrimeScript Ex Taq Ⅱ (TaKaRa), and sense and antisense primers in a final reaction volume of 20 μL using LightCycler 2.0 (Roche, Basel, Switzerland). The primer sequence used for the detection of CVB3 RNA was 5′-GCACACACCCTCAAACCAGA-3′ (sense) and 5′-ATGAAACACGGACACCCAAAG-3′ (antisense). PCR cycling condition started at 94 ℃ for 1 min, followed by annealing at 55 ℃ for 2 min and the procedure of extension at 72 ℃ for 3 min. 30 cycle was used. GAPDH mRNA was used as internal control for quantifying viral RNA. The 2-ΔΔCt method was used to calculate the relative levels of viral RNA (Livak and Schmittgen 2001).
-
Viral titers were determined by the plaque assay. Briefly, the viral stock was serially diluted with maintenance medium. HeLa cells were seeded in six-well plates at the density of 2 × 105 cells/well and incubated for 18–24 h at 37 ℃ with 5% CO2. When the cell culture reached ~90% confluence, cells were washed with PBS and overlaid with 450 μL of viral diluent. The cells were incubated with medium containing virus for 1 h to allow the adhesion of the virus to the cells, and the supernatant was then removed. Finally, the cells were overlaid with 2 mL of DMEM medium containing 5% FBS and 0.8% agarose. The culture plates were incubated in a humidified chamber for 30 min and then placed in an inverted position. Cells were incubated for another 72 h at 37 ℃ with 5% CO2 before being stained with 0.05% neutral red (Sigma, St. Louis, MO, USA) for 1 h after which plaques were counted and viral titers (pfu/mL) were calculated.
Cell Lines and Viruses
Plasmid Construction
Transfection
Fluorescence Observation
Western Blotting
Cell Viability Assay
Quantitative Reverse Transcription PCR (RTqPCR)
Plaque Forming Assay
-
To study the effect of CVB3 infection on SG formation, we first constructed a cell line HeLaEGFP-TIA1 that stably expressed TIA1, a well-documented constituent of SGs. We then observed the expression and localization of TIA1 in HeLaEGFP-TIA1 cells after CVB3 infection. HeLaEGFP-TIA1 cells were mock infected with DMEM or infected with CVB3 at an MOI of 10 or 50, respectively. Starting at 4 h of post-infection (p.i.), the distribution of EGFP-TIA1 displayed an obvious granular pattern in the cytoplasm of CVB3-infected cells (Fig. 1).
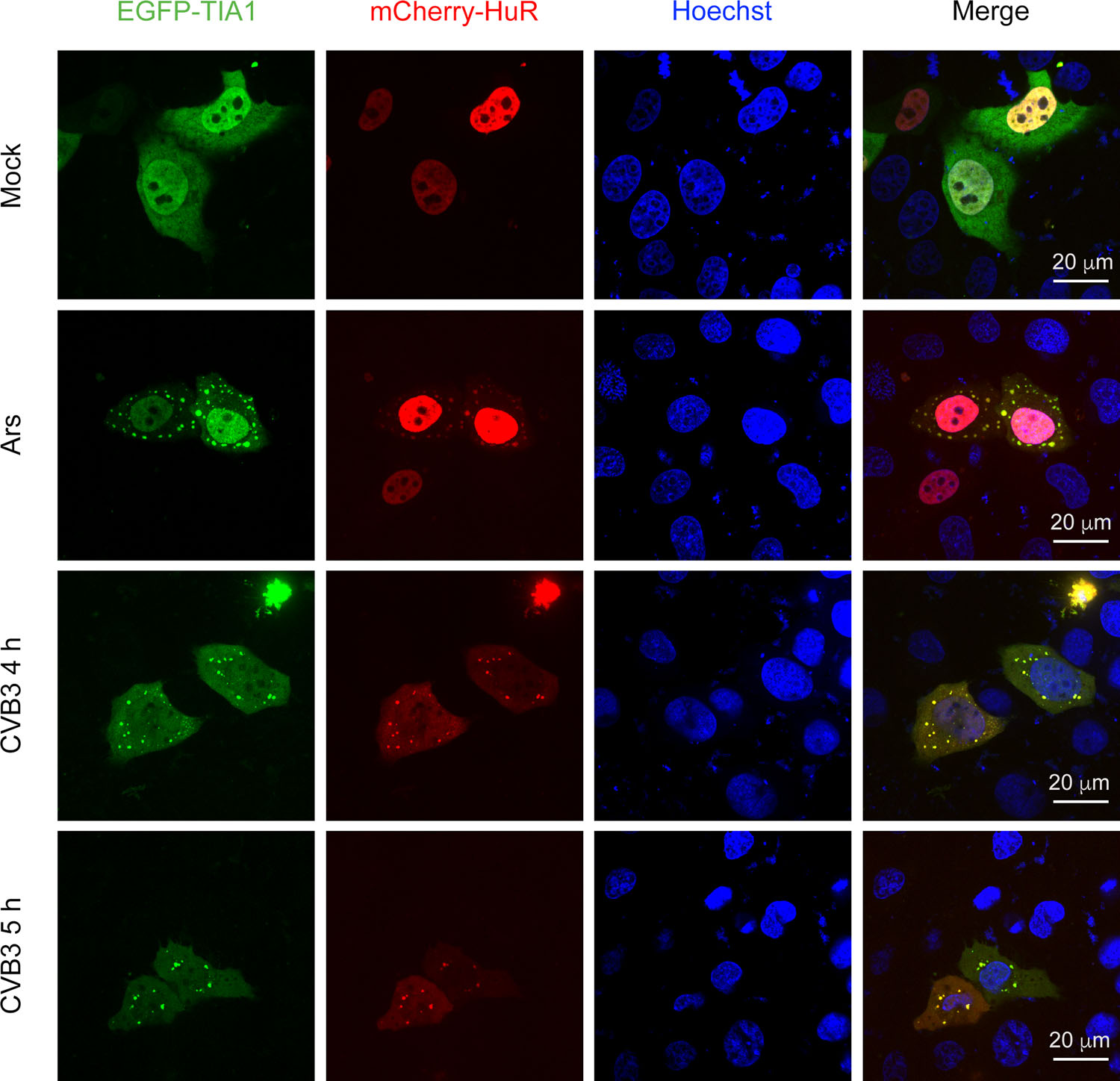
Figure 1. Co-localization of TIA1 and HuR in the cytoplasmic granules of CVB3-infected cells. HeLaEGFP-TIA1 cells were infected with CVB3 (MOI = 10) for 4 and 5 h, respectively. Control cells were treated with Ars. Cell nuclei were stained with Hoechst 33258. The expression and distribution of EGFP-TIA1 and mCherry-HuR were observed with a fluorescence microscope.
To further verify that CVB3 infection could induce SG formation, we examined the co-localization of EGFP-TIA1 and mCherry-HuR during CVB3 infection. mCherry-HuR was distributed predominantly in the nucleus of mock-infected cells, but it re-localized to cytoplasmic granules during the CVB3 infection (Fig. 1, second column from left). Furthermore, EGFP-TIA1-positive granules were colocalized with mCherry-HuR-positive granules (Fig. 1, fourth column from left) in cells infected with CVB3. We also noticed that SGs containing TIA1 and HuR did not contain G3BP1 at 6 h p.i (data not shown). Similar results were obtained in cells infected with CVB3 at an MOI of 50 (data not shown). Taken together, these results demonstrated that CVB3 infection could induce the formation of SGs, which might contain distinct protein contents compared with SGs induced by oxidative stress.
-
To evaluate the effect of CVB3 replication on the formation of SGs, HeLaEGFP-TIA1 cells were treated with 0.5 mmol/L Ars for 1 h and then infected with CVB3 for 3, 4, 5, 6, and 7 h. Control cells were treated with Ars only. The formation of SGs was evaluated by observing the expression and distribution of EGFP-TIA1 under a fluorescence microscope. SGs appeared in cells treated with Ars. CVB3 infection did not lead to a significant change in SG size or density compared with SGs in cells treated with Ars alone (Fig. 2A). This observation indicates that CVB3 infection did not affect the pattern of SG formation induced by Ars.
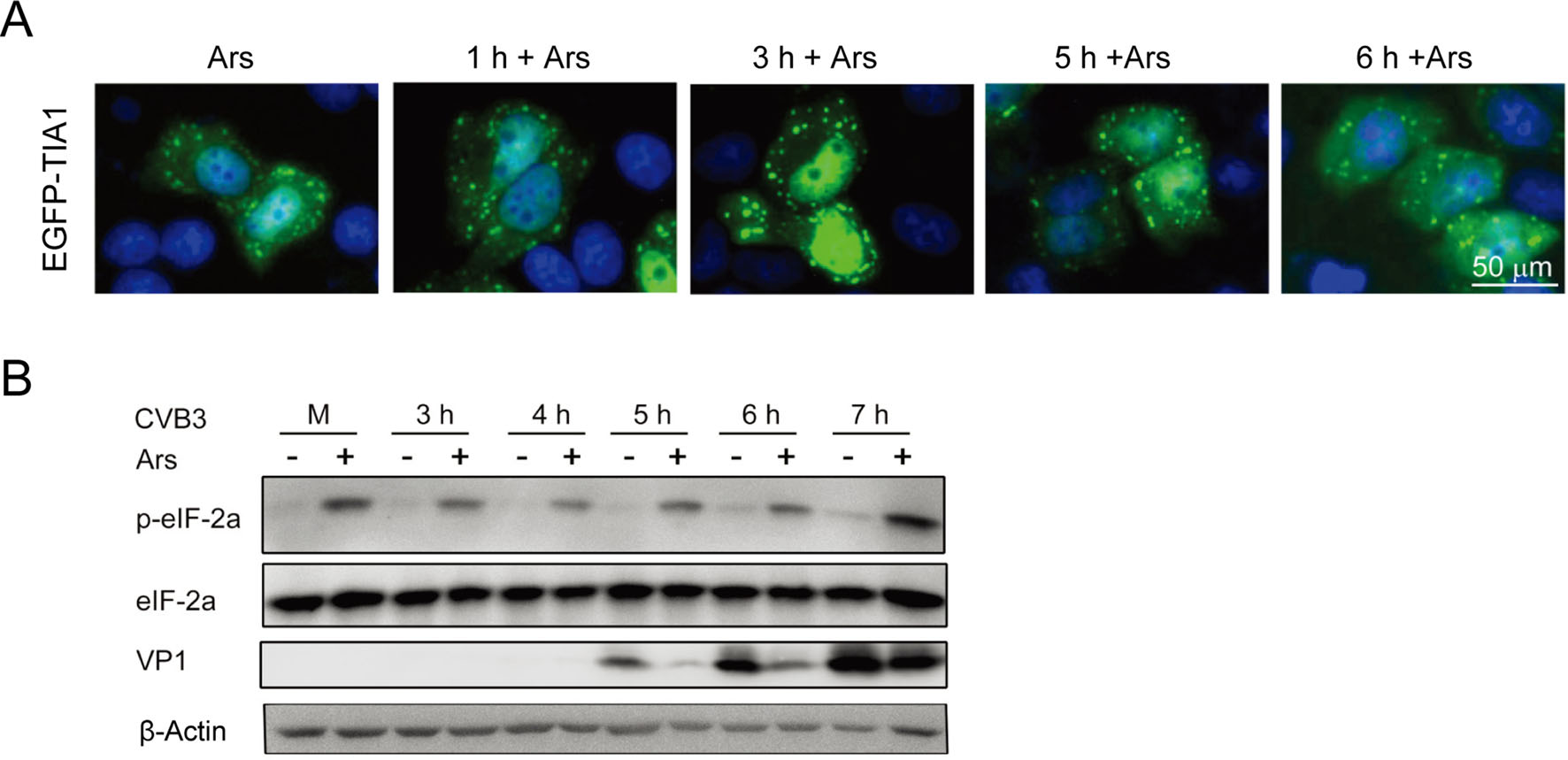
Figure 2. SGs induced by Ars affect the biosynthesis of CVB3. A HeLaEGFP-TIA1 cells were treated with 0.5 mmol/L Ars for 1 h and infected with CVB3 for various time periods (from 1 h to 6 h). Control cells were treated with Ars only. The formation of SGs was evaluated by observing the expression and distribution of EGFP-TIA1 with a fluorescence microscope. B HeLa cells were treated with 0.5 mmol/L Ars for 1 h and then infected with CVB3 for 3, 4, 5, 6, 7 h. Cells were harvested and cellular proteins were subjected to the analysis of western blotting. Mock-infected cells were treated with DMEM. Cells infected with CVB3 without Ars treatment were used as controls.
To explore the role of SGs in the biosynthesis of CVB3, HeLa cells were treated with Ars for 1 h and then infected with CVB3 for 7 h. Cellular proteins were analyzed with Western blotting at various time points of viral infection. Mock-infected cells were treated with DMEM. Control cells were infected with CVB3. Both CVB3 infection and Ars treatment had no effect on the level of eIF-2α compared with controls (Fig. 2B). However, the levels of phosphorylated eIF2α (p-eIF2α) were significantly increased in all virus-infected cells treated with Ars, while no p-eIF2α was detected in mock-infected cells or in CVB3-infected cells not treated with Ars. Low levels of p-eIF2α were detected at 6 and 7 h after CVB3 infection. Moreover, when treated with Ars, the levels of VP1 were significantly reduced at 5, 6, and 7 h p.i., compared with that in cells not treated with Ars. These results indicate that the pre-formed SGs (induced by Ars) in the host cells inhibit the biosynthesis of CVB3, especially at the early stage after the initiation of viral infection.
-
Studies have reported that overexpression of cleavage-resistant G3BP1 inhibited the production of poliovirus progeny (White and Lloyd 2011). In the current study, we first evaluated the efficiency of si-G3BP1. HeLa cells were co-transfected with pEGFP-G3BP1 and si-G3BP1, while control cells were cotransfected with pEGFP-G3BP1 and scrambled siRNA (siControl). The efficiency of si-TIA1 was evaluated in the same manner. At 24, 48, and 72 h p.i., the expression of green fluorescence was observed with fluorescence microscopy. Cell proteins were then extracted and the concentration of green fluorescent protein was determined. The intensity of green fluorescence of cells transfected with either si-G3BP1 or siTIA1 was significantly reduced, compared with that in the cells treated with control siRNA at 48 h p.i. (Fig. 3A; data obtained at 24 and 72 h p.i were similar). At 48 h after transfection, the fluorescence intensity in the cells transfected with si-G3BP1 was reduced to less than 40% relative to that of the control cells. Similarly, cells transfected with si-TIA1 showed about 30% fluorescence intensity relative to that of the control cells (Fig. 3A–b, Fig. 3A–d). The viability of cells transfected with siRNAs was determined at 48 h p.i. using the MTT assay. The results showed that cell viability was not affected by the transfection of si-G3BP1 or si-TIA1 (Fig. 3B).
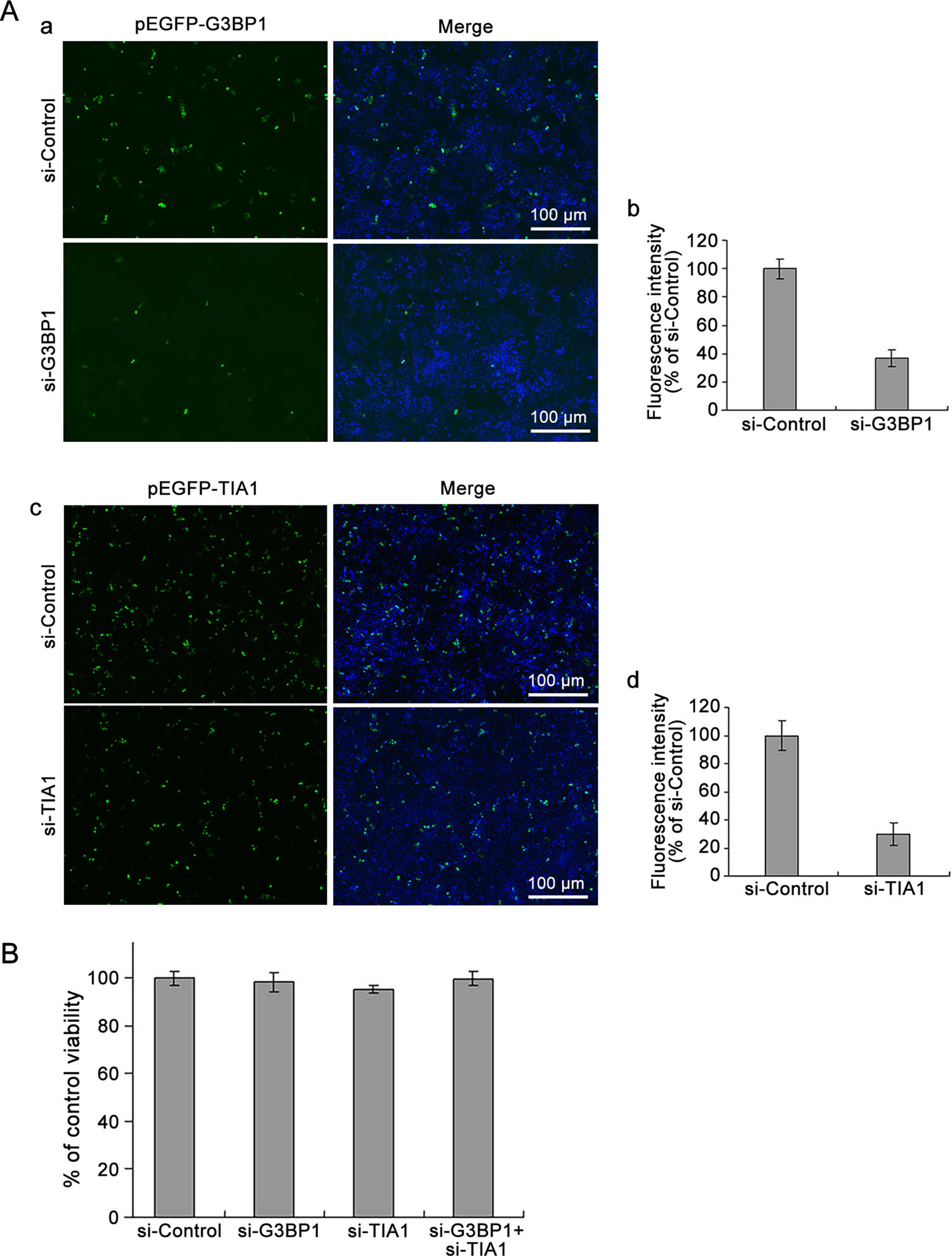
Figure 3. Knockdown of G3BP1 and/or TIA1 does not affect cell viability. A HeLa cells were cotransfected with pEGFP-G3BP1 and si-G3BP1 or pEGFP-TIA1 and si-TIA1 for 48 h. Control cells were co-transfected with pEGFP-G3BP1 and si-Control or pEGFP-TIA1 and si-Control (a and c). The expression of EGFP-G3BP1 and EGFP-TIA1 were determined with a fluorescence microscope (a and c). Fluorescence intensity was determined with a NanoDrop 3300 (b and d). B HeLa cells were transfected with either siG3BP1 or si-TIA1, or siG3BP1 and si-TIA1 for 48 h. Control cells were treated with siControl. Cell viability was determined using the MTT assay (n = 4).
To study the effect of si-G3BP1 and/or si-TIA1 on CVB3 biosynthesis, cells were transfected with si-G3BP1, si-TIA1 or co-transfected with si-TIA1 and si-G3BP1 for 24 h and were then infected with EGFP-CVB3. Figure 4A and 4B show the results of cells infected with EGFP-CVB3 at 22 and 44 h p.i., respectively. At 44 h p.i., the intensity of green fluorescence was significantly increased in cells transfected with either si-G3BP1 or si-TIA1, while cells co-transfected with si-G3BP1 and si-TIA1 showed even higher increases compared with the control cells (Fig. 4B). Cellular proteins were then harvested at 44 h of p.i. and viral VP1 protein was detected by western blotting. The VP1 level was increased in cells transfected with either si-G3BP1 or si-TIA1, or siG3BP1 and si-TIA1, indicating that si-G3BP1 and/or si-TIA1 could promote the protein biosynthesis of CVB3 (Fig. 4C). Collectively, these data suggest that the formation of SGs might play an inhibitory role in the biosynthesis of CVB3.

Figure 4. Knockdown of G3BP1 and/or TIA1 enhances the biosynthesis of EGFP-CVB3. HeLa cells were transfected with si-Control, siG3BP, si-TIA1, and si-G3BP1 and si-TIA1, respectively, for 24 h. Cells were then infected with EGFP-CVB3 (MOI = 0.01) for 44 h. EGFP expression observed with a fluorescence microscope at A 22 h p.i. and B 44 h p.i.. C The expression of VP1 at 44 h p.i. was detected by western blotting.
-
To further confirm the foregoing results, the effect of siG3BP1 and/or si-TIA1 on the biosynthesis of CVB3 Woodruff strain was determined. Cells were transfected with siG3BP1 and/or si-TIA1 for 24 h and then infected with CVB3. Cytopathic effects (CPE) were observed at 40 h p.i.. CPE in cells transfected with si-G3BP1 and si-TIA1 was significantly increased at 40 h p.i., compared with cells transfected with control siRNA. Cells transfected with either si-G3BP1 or si-TIA1 also showed increased CPE compared with controls (Fig. 5A). At 44 h after CVB3 infection, viral RNA was determined with RT-qPCR. Viral RNA was increased in cells transfected with either si-G3BP1 or siTIA1, or si-G3BP1 and si-TIA1, compared with that in the control cells (Fig. 5B). At 44 h p.i., VP1 expression was also increased in cells transfected with either si-G3BP1 or siTIA1, or si-G3BP1 and si-TIA1 (Fig. 5C).
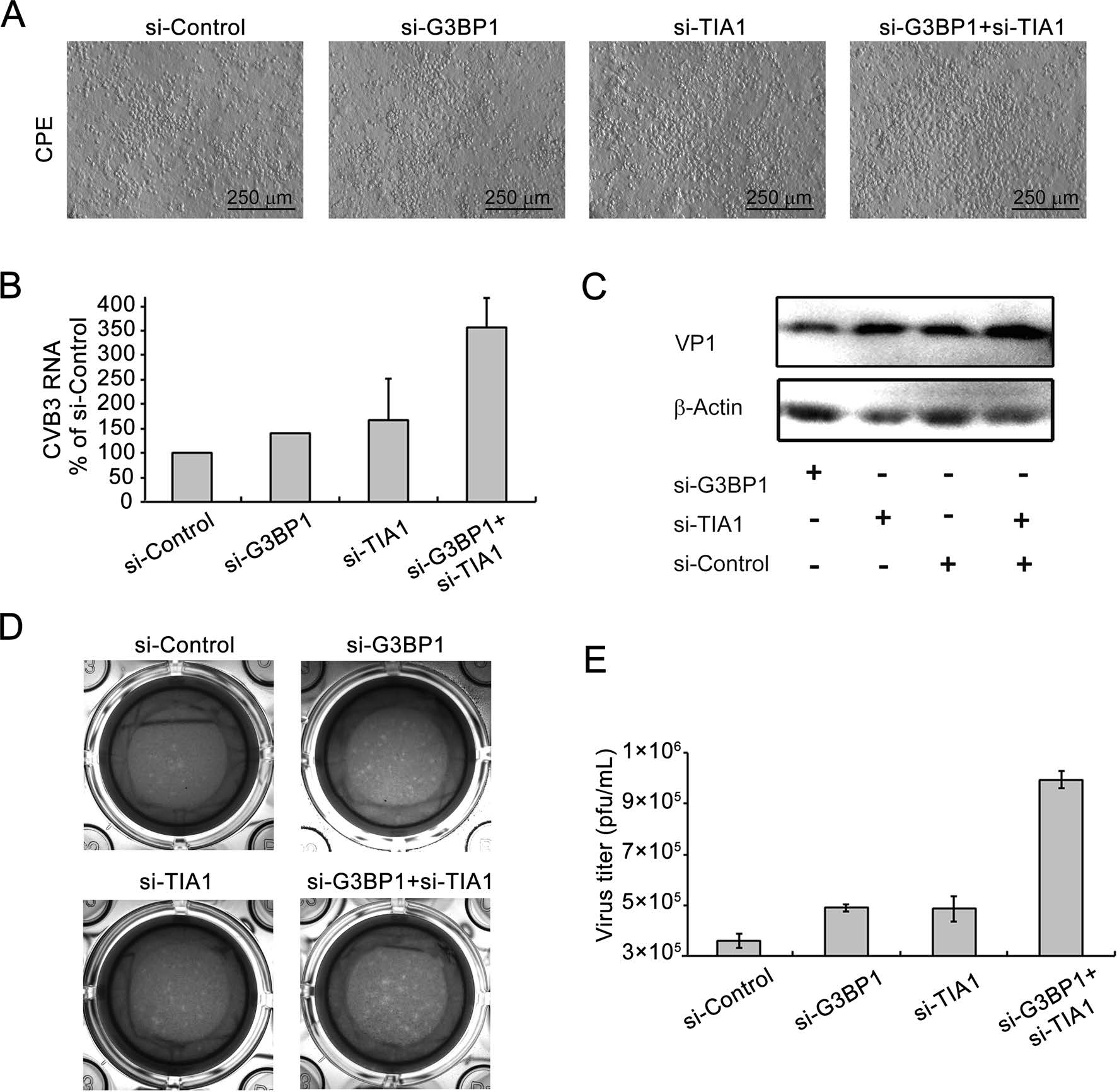
Figure 5. Knockdown of G3BP1 and/or TIA1 enhances the biosynthesis of CVB3 wild type. A HeLa cells were transfected with si-G3BP1 and/or si-TIA1 for 24 h and infected with CVB3 (MOI = 0.01) for 40 h and CPE were observed. B Cells were treated with siRNAs as described in (A) and infected with CVB3 (MOI = 0.01) for 44 h. Total RNA was extracted and viral RNA was determined using RTqPCR. C Cell lysate was subjected to Western blot analysis. D–E CVB3 titers were measured using the plaque formation assay. (n = 4). * P < 0.05; ** P < 0.01.
CVB3 titers were measured using the plaque formation assay by collecting the supernatant of culture media at 44 h of p.i.. The number of the plaques that formed by cells transfected with si-G3BP1 and si-TIA1 increased significantly compared with control (Fig. 5D, 5E). Transfection with either si-G3BP1 or si-TIA1 alone also led to increased numbers of plaques. These results show that knocking down G3BP1 and/or TIA1 promotes the biosynthesis of CVB3, suggesting that SGs may play an antiviral role during CVB3 infection.
CVB3 Induces SGs Formation in Infected Cells
Ars-Induced SGs Inhibit CVB3 Biosynthesis
Suppressed SG Formation Promotes the Biosynthesis of EGFP-CVB3
Suppressed Formation of SGs Enhances the Biosynthesis of Wild Type CVB3
-
The formation of SGs is a significant cellular process that occurs when cells are exposed to external stimuli. Viruses can interact with SGs in a variety of ways (Miller 2011; Panas et al. 2012; Garaigorta et al. 2012; Mok et al. 2012). The current study aimed to understand how SG formation could affect CVB3 replication.
Studies have shown that SGs act as a part of the antiviral host response in a variety of viral infections. SGs can inhibit the replication of mouse hepatitis virus (Raaben et al. 2007), influenza A virus (Mok et al. 2012), and reovirus (Choudhury et al. 2017) to a certain extent. In the current study, G3BP1, TIA1, and HuR were used as marker proteins for the detection of SGs. TIA1 is located in a variety of stimulus-induced SGs, although it has been reported that TIA1 overexpression has no significant effect on the replication of poliovirus (White and Lloyd 2011). In the current study, we first generated the cell line HeLaEGFP-TIA1, which stably expressed EGFP-TIA1. Using this cell line, the formation of SGs could be observed with a fluorescence microscope in real time, and the infection of CVB3 could be quantitatively determined.
By observing the co-localization of TIA1 and HuR in CVB3-infected cells, we found that CVB3 could effectively induce SG formation and that TIA1 and HuR molecules were localized in SGs at the early stage of viral infection. Interestingly, at later stages of CVB3 infection, G3BP1 and eIF4G were not localized in SGs (data not shown). Our results were similar to the reported characteristics of SGs caused by poliovirus (White and Lloyd 2011; Dougherty et al. 2011, 2015; Lloyd 2016). At later stages of poliovirus infection, G3BP1 and eIF4G were not localized in SGs, as a result of the proteolytic cleavage caused by viral 3C protease (White and Lloyd 2011; Dougherty et al. 2011, 2015; Lloyd 2016). A similar phenomenon was also found during the infection of enterovirus 71 (Wu et al. 2014a, b). These findings indicate that enteroviruses share similar features regarding the induction of SGs. In this study, SG formation was observed at 3 h after CVB3 infection. In contrast, SGs were observed at 2 h after infection using the same amount of poliovirus (Chapman et al. 2000; Harkins et al. 2005). This difference might be related to the fact that the growth cycle of CVB3 is slightly longer than that of poliovirus (Chapman et al. 2000; Harkins et al. 2005).
To explore the role of SGs in the biosynthesis of CVB3, the effect of Ars-induced SGs on viral replication was determined. We found that CVB3 VP1 levels were decreased in the cells treated with Ars, suggesting that the formation of SGs induced by Ars could limit viral replication to certain extent.
Arshasalsobeenshowntohaveothereffectsoncells, such asalteringthecellcycle(Montero andTrujillo-Alonso 2011). Indeed, suppressed expression of the SG constituent proteins G3BP1 or TIA1 significantly inhibited SG formation (Panas et al. 2012; White and Lloyd 2011). For this reason, we used siRNAs to observe the effect of SG related proteins on CVB3 biosynthesis. When the expression of either G3BP1 or TIA1 alone or in combination was inhibited by siRNA, CVB3 replicationlevelswereincreased.ThesedatasuggestthatSGs may restrict viral replication. The formation of SGs may be part of the antiviral response of the host cell. It has been reported that G3BP1 overexpression restored SG formation in cells infected with poliovirus, while viral replication levels were reduced by nearly one-third (White and Lloyd 2011; Dougherty et al. 2011, 2015). These data support our speculation that SGs have an antiviral role in the infection of enteroviruses.
Viral infection can cause eIF2α phosphorylation, although this does not necessarily lead to SG formation (Slaine et al. 2017; Amorim et al. 2017). Our results showed that CVB3 infection did not affect the formation of Ars-induced SGs, but that the levels of p-eIF2α in CVB3-infected cells treated with Ars were significantly increased, possibly due to a mechanism associated with prolonged treatment of Ars which promotes eIF2α phosphorylation.
In summary, this study demonstrated that CVB3 infection could effectively induce SG formation, while SGs could in turn restrict the biosynthesis of CVB3 in host cells. Our observation suggests that SGs might have an antiviral role during CVB3 infection, especially during the early stage of viral infection. These findings provide new insight for understanding the interactions between CVB3 and the host cell.
-
This study was supported by the Natural Science Foundation of China (Grant 81571999 to Z Zhong; 81672007 to W Zhao; 81772188 to Y Wang, 31300144 to T Wang). We are grateful to the technical support from Heilongjiang Provincial Key Laboratory of Pathogens and Immunity and Heilongjiang Provincial Science and Technology Innovation Team in Higher Education Institutes for Infection and Immunity of Harbin Medical University.
-
ZZ designed and guided the study. XZ, SW, LL, TW, XZ, YC, and WX performed the experiments. LT and YW contributed in the collection and analysis of data. ZZ and WZ wrote the manuscript. All authors read and approved the final manuscript.
-
The authors declare that they have no conflict of interest.
-
This article does not contain any studies with human or animal subjects performed by any of the authors.

 DownLoad:
DownLoad: