














HTML
-
Seneca Valley virus (SVV) is the only member of the genus Senecavirus in the family Picornaviridae (Knowles and Hallenbeck 2005; Hales et al. 2008). SVV is highly related to the Cardiovirus genus and is also different with other picornaviruses. The genome of SVV is a singlestranded, nonsegmented, positive-sense RNA, with length of about 7.3 kb (Hales et al. 2008). The viral genome contains an open reading frame (ORF) encoding a large polyprotein that is subsequently processed into various mature viral proteins. The ORF was divided into the L, P1, P2, and P3 regions. The L region encodes the leader protein (Lpro) that self-cleaves from the nascent viral polyprotein precursor. The P1 region is defined as that part of the polyprotein that generates the four structural proteins (VP1, VP2, VP3, and VP4), and the P2 and P3 regions produce viral nonstructural proteins (2A, 2B, 2C, 3A, 3B, 3C, and 3D) (Leme et al. 2017).
The first SVV strain was incidentally discovered in contaminated cell cultures in 2002 in the United States (US), and was initially used for the treatment of neuroendocrine tumors in humans due to its potential oncolytic activity (Reddy et al. 2007). SVV was reported to be associated with sporadic cases of vesicular disease in pigs (Pasma et al. 2008; Leme et al. 2017). In recent years, different isolates of SVV have been reported in the outbreaks of vesicular disease outside of North America, including Brazil, Colombia, China, and Thailand (Leme et al. 2016; Saeng-Chuto et al. 2017; Sun et al. 2017; Zhu et al. 2017). The SVV strains isolated in Guangdong Province often cause apparent diseases in sows and piglets. The clinical symptoms include fever, anorexia, lethargy, and lameness. Vesicular lesions are found in the hoofs and mouth. Several newborn piglets exhibit acute death. The foot-and-mouth disease virus (FMDV), swine vesicular disease virus (SVDV), vesicular exanthema of swine virus (VESV), and vesicular stomatitis virus (VSV) were tested negative in those SVV-positive samples (Wu et al. 2016).
The VP1 phylogenetic tree showed that the SVV strains isolated from Guangdong were distant from the historical SVV prototype, SVV-001 strain (92.4%–92.8% nucleotide identity) (Zhao et al. 2017). Hubei is the second province in China that has reported outbreaks of SVV infection in pig farms. The reported SVV strain isolated in Hubei Province in 2016 was most closely related to the Guangdong strains isolated in 2015, which implies that the Hubei strain might originate from Guangdong (Qian et al. 2016).
In January 2017, in Fujian and Henan provinces in China, outbreaks of SVV infection were reported. Three new SVV isolates, CH-FJ-2017, CH-HN-2017 and CH-HNSL-2017 were identified and the viral genomes were sequenced. The three isolates had the highest polyprotein amino acid identity (99.5%–99.6%) to the US SVV strain, KS15-01, whereas, they only shared 97.2%–98.9% polyprotein amino acid identity with previous Chinese SVV strains. The phylogenetic analysis also clustered the three strains into the same branch with KS15-01, suggesting that the SVV distribution in China is complicated (Zhu et al. 2017).
In March 2017, porcine idiopathic vesicular disease (PIVD) occurred in two different pig farms in Guangdong Province. Vesicles and ulcerative lesions were found on the snout and hooves of the affected pigs. The samples from the pigs that had fluid-filled vesicles or ulcerative lesions on the coronary band were tested for SVV, FMDV, SVDV, VESV, and VSV. The viral genomes were determined and phylogenetic analyses were performed to contribute to a better understanding of the emergence, spread, and evolution of the identifed viruses.
-
The fluid-filled vesicles and ulcer tissue samples from the affected pigs in two pig farms in Guangdong Province were collected in March, 2017. Baby hamster kidney cells (BHK21 cell line) were provided by the Lanzhou Veterinary Research Institute and used for virus isolation. The cells were cultured in minimum essential medium (MEM) supplemented with 10% fetal bovine serum (FBS) at 37 ℃. Competence trans5α and pMD20-T vectors were purchased from the Takara (Dalian, China) Company.
-
Fetal bovine serum (FBS), trypsin and minimum essential medium (MEM) were purchased from the HyClone Laboratories (Logan, Utah, USA). TRIzol, LA Taq polymerase, PrimeSTAR® HS DNA polymerase, M-MLV reverse transcriptase, random primers (6-mer), DNA A-Tailing Kit, DNA marker, Oligo dT primer, and T4 DNA ligase were purchased from the Takara Company (Dalian). Agarose was purchased from the BIOWEST Company (Biowest, Spain). The One-step RT-PCR kit, E.Z.N.A.® Plasmid Mini Kit I and Agarose Gel DNA extraction kit were purchased from the Omega Company (Norcross, GA, USA).
-
Vesicles and ulcerative tissue were collected, mixed and milled with phosphate buffered saline (PBS) at 1:10 to generate homogenates. The mixtures were frozen and thawed repeatedly for three times, and centrifuged for 10 min at 10, 000 rpm, 4 ℃ to collect supernatants. Total RNA was extracted from the supernatants using RNA TRIzol reagent according to the manufacturer's instructions. The first strand of cDNA was synthesized using M-MLV reverse transcriptase. SVV, SVDV, VSV, VESV and FMDV can cause similar vesicular diseases. Therefore, the synthesized cDNAs were used for the detection of those viruses. The primers and detection methods were described previously (Bao et al. 2008; Zheng et al. 2015; Bracht et al. 2016; Zhao et al. 2017).
-
The prepared supernatant specimens with SVV PCR positive results were filtered through 0.45 μm filter and used to inoculate the confluent monolayer BHK-21 cells. Mockinfected BHK-21 cells were used as the negative control. After inoculation, the cells were cultured at 37 ℃ with 5% CO2 for adsorption. Following 1 h of adsorption, the inoculum was replaced by DMEM with 1% fetal bovine serum (FBS). The cytopathic effect (CPE) was observed and recorded every day. After four continuous passages, the cells and medium were collected. The cultures were frozen and thawed repeatedly three times and centrifuged for 10 min at 1000 rpm and 4 ℃ to remove cell debris.
-
The complete SVV VP1 genes were amplified using the specific primers from the synthesized cDNA as described previously (Bracht et al. 2016). The PCR system (20 μL) included: LA Taq Polymerase premix (10 μL), cDNA (2.4 μL), forward primer and reverse primer (0.4 μL each), and nuclease free water to a total volume of 20 μL. The amplification parameters were set as follows: 95 ℃ initial denaturation for 5 min; 32 cycles of 95 ℃, denaturation for 1 min, annealing at 57 ℃ for 30 s, extension at 72 ℃ for 2 min; final extension for 10 min at 72 ℃ and 4 ℃ hold. The amplified fragments were cloned into the pMD20T vector and submitted to the Beijing Genomics Institute (BGI) Company for gene sequencing. The sequencing data were analyzed by BLAST in the GenBank of NCBI.
-
Eight pairs of primers referring to the previously published SVV sequences in NCBI GenBank were designed and used for amplification of the viral genome (Supplementary Table S1). The primers were synthesized by the BGI Company. The amplified PCR fragments were cloned into the pMD-20T vector and sequenced by the BGI Company. The SVV gene sequences of each fragment were analyzed and assembled using the DNAstar software, and the complete genome sequences of the viruses were subjected to BLAST analysis.
-
All the complete genome sequences of SVV available in the NCBI GenBank until November, 30th, 2017 were downloaded and used for the genetic and phylogenetic analyses. The phylogenetic tree was constructed by MEGA 6 software. Phylogenetic analyses of the complete genomes were performed using the neighbor-joining method and selection of Kimura-2-parameter nucleotide substitution model. The bootstrap replicates were set as 1000.
Specimens, Cells and Vectors
Reagents
RNA Extraction and Real-Time PCR
Virus Isolation
The VP1 Gene Detection
Virus Genome Cloning and Sequencing
Genetic and Phylogenetic Analyses
-
The affected pigs exhibited the clinical signs of vesicular disease. Thus the agents of pig vesicular disease, including FMDV, SVV, SVDV, VESV and VSV were tested. All the viruses were tested negative, except for SVV. The filtered specimens were inoculated onto the confluent monolayer BHK21 cells to isolate the virus. At 12 h postinfection, CPE, characterized by cell rounding, disruption and focal detachment, was observed in virus-infected cells, but not in mock-infected cells (Supplementary Figure S1A). The virus specimens were serially propagated in cultured BHK21 cells, collected, and subjected to RT-PCR detection with primers specific to the SVV VP1 gene. As shown in Supplementary Figure S1B, the amplified products were 975 bp in length. The amplified fragments were subsequently cloned into the pMD-20T vector, and the constructed plasmids were sent to BGI Company for sequencing. Sequence analysis was carried out using the BLAST program of GenBank (NCBI). The analyses indicated that the two amplified VP1 gene sequences showed 98.9% and 99.1% nucleotide similarity with the SVV USA/GBI29/2015 strain (KT827251). Therefore, we successfully isolated two SVV strains and named them SVV CH-GD-2017-1 and SVV CH-GD-2017-2.
-
The viral genome sequences of CH-GD-2017-1 and CH-GD-2017-2 were further determined to evaluate the characteristics of the newly isolated viruses. Eight pairs of primers were designed with Primer Premier 5.0 software (Premier Biosoft International, Palo Alto, CA, USA) and used for amplification of the different regions of the SVV genome. The expected fragments were amplified by PCR and subjected to sequencing and analysis (Supplementary Figure S2). The sequences were assembled using DNAstar software. The complete genome sequences were submitted to GenBank. The results showed that the lengths of the two SVV were 7285 nt (SVV CH-GD-2017-1) and 7284 nt (SVV CH-GD-2017-2). The accession numbers were SVV CH-GD-2017-1 (MF189000) and SVV CH-GD-2017-2 (MF189001), and the viral genome both contained the same genome organization: 5'-UTR-L-VP4-VP2-VP3-VP1-2A-2B-2C-3A-3B-3C-3D-3'-UTR, without insertions or deletions. The length of each gene of the viral genome was shown in Table 1.

Table 1. The genome organization of SVV CH-GD-2017-1 and SVV CH-GD-2017-2, as well as the sequence identity of each gene with other strains available in GenBank.
-
The nucleotide sequences of the two isolated strains were compared to the other 52 SVV strains (available in the GenBank). We determined that both CH-GD-2017-1 and CH-GD-2017-2 showed high genomic nucleotide similarity with the SVV strains isolated from the US in 2015–2016, and had the highest similarity with the US strain USA/ IA39812/2015_P1 (KU95408). CH-GD-2017-1 showed 98.6% nucleotide similarity with USA/IA39812/2015_P1, and CH-GD-2017-2 showed 98.8% with USA/IA39812/ 2015_P1. The two isolates showed 97.7%–98.8% nucleotide similarity with other recent US strains, and shared 96.1%–98.5% nucleotide similarity with other Chinese strains. Both CH-GD-2017-1 and CH-GD-2017-2 were more closely related to the US strains than previous Chinese strains.
Comparisons of the amino acid similarities of different genes from CH-GD-2017-1 and CH-GD-2017-2 to other SVV strains were also performed (Table 1). We found that the VP4 proteins of all other strains had 100% amino acid similarity except for the two Thailand strains, CH-GD-2017-1 and CH-GD-2017-2. Compared to the other SVV strains, the VP4 of CH-GD-2017-1 and CH-GD-2017-2 included a unique mutation at amino acid 48 (N48S, from asparagine to serine). The two Thailand strains included a unique mutation at amino acid 65 (N65S). Thus, new mutations might have appeared in the recent SVV strains isolated in China and Thailand. In addition, we observed that CH-GD-2017-1 and CH-GD-2017-2 shared 100% amino acid similarity in 3B with all other SVV strains except SVA CH/GXI09/2016. Alignment of the amino acid sequences of all SVV strains suggested that the VP4, VP2, 2B, and 2C were relatively conservative. The 2C protein was the most highly conserved protein in all SVV strains (98.8%–100% similarity), while 3A was the most variable protein in all SVV strains (90%–98.9% similarity). Therefore, the 3A gene could be used as a target gene to evaluate SVV variation and diversity.
-
A phylogenetic tree for the SVV strains was constructed using complete genome sequences. The results indicated that CH-GD-2017-1 and CH-GD-2017-2 were clustered in a branch with all US SVV strains, as well as the Chinese strains isolated in 2017 (also included one strain isolated in December 2016) (Fig. 1). The CH-GD-2017-1 and CH-GD-2017-2 were most closely related to the US USA/ IA39812/2015_P1 strain, and showed a distant relationship to Chinese strains isolated before December 2016, as well as all previous Guangdong strains. The Chinese strains isolated before 2017 except the SVA/HLJ/CHA/2016 strain (Isolated in December 2016) (Wang et al. 2017) were more closely related to the Brazilian or Canadian strains.
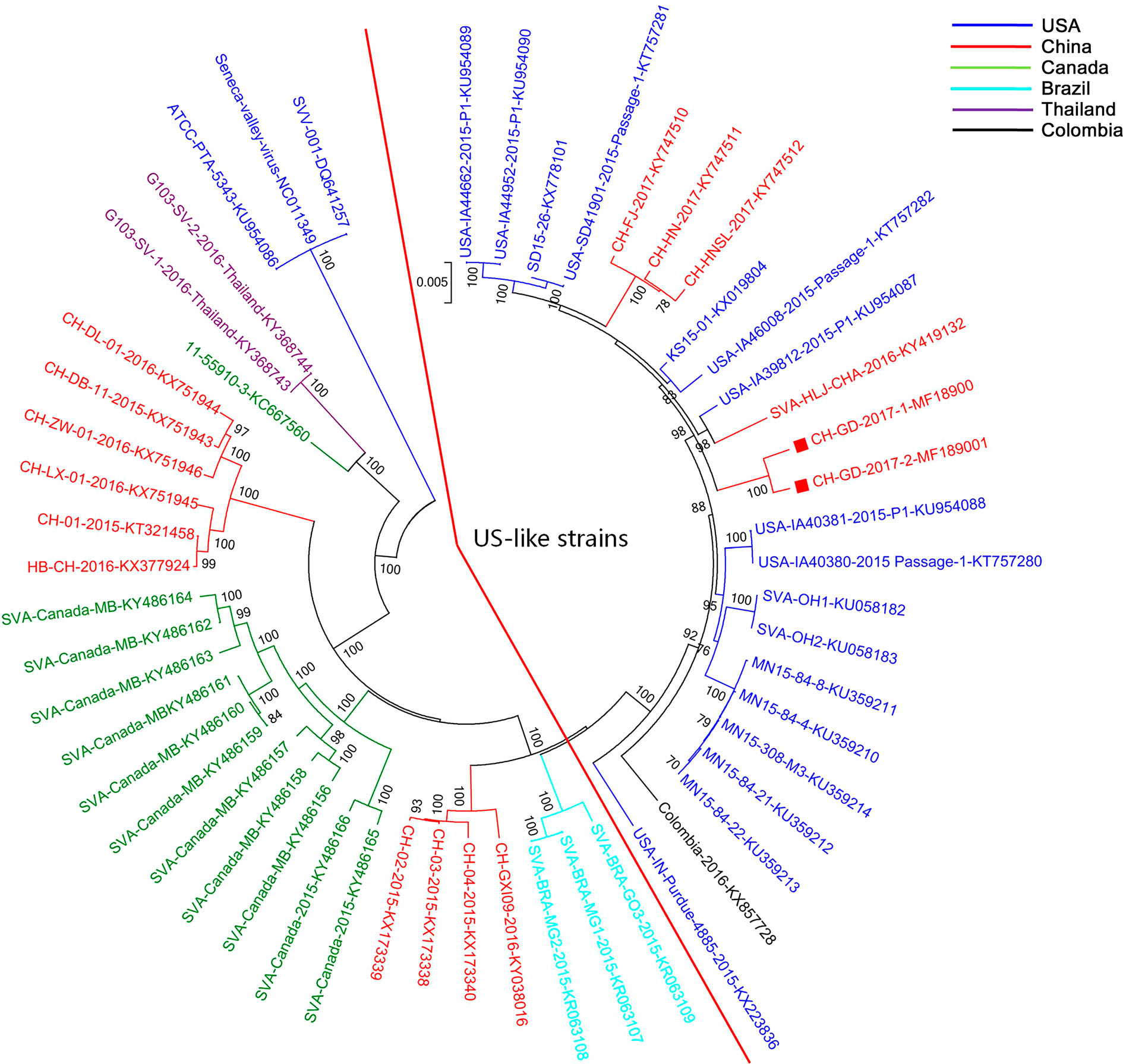
Figure 1. Phylogenetic analyses of all SVV strains available in the GenBank.
-
We also compared the amino acids mutations in CH-GD-2017-1 and CH-GD-2017-2 to the first identified SVV strain, SVV-001, and the first reported Chinese strain, SVV CH-01-2015. As shown in Table 2, there were 55 mutated amino acids between the new strains and SVV-001, and 28 mutated amino acids between the new strains and SVV CH-01-2015. The 3A, VP3 and 3D proteins included more mutated amino acids than the other viral proteins, with 3A being the most variable viral protein.

Table 2. Mutation analysis of the amino acid sequences of CH-GD-2017-1 and CH-GD-2017-2, compared to SVV CH-01-2015 and SVV-001 strains.
-
SVV was first identified in 2002. In 2007, SVV was detected in swine. SVV was only reported in the US and Canada before 2014, while it was reported in additional countries after 2015. We analyzed reports of SVV strains from different countries based on the genome sequences available in GenBank at the time of writing this article. At present, 20 strains have been reported in the US, 16 in China, 12 in Canada, three in Brazil, one in Thailand, and one in Colombia (Fig. 2). The US and China have reported most of the SVV cases, and no cases have been reported in European and African countries. In China, SVV cases mainly emerged in the eastern and central regions, with no cases being reported in inner China (Fig. 3). Based on the isolation date recorded in GenBank, SVV outbreaks mainly occurred during the winter and spring.
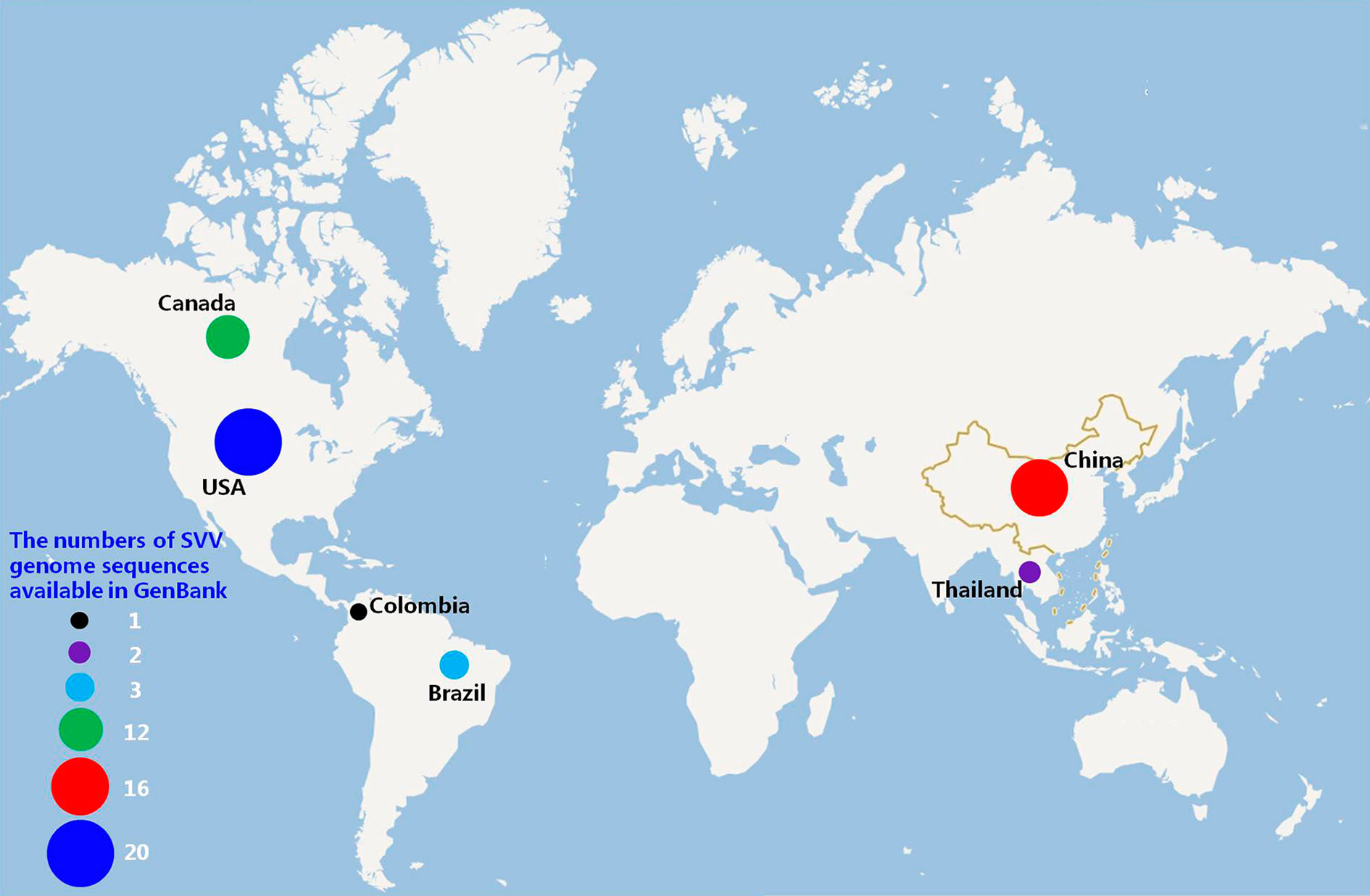
Figure 2. Global status and geographical distribution of SVV.
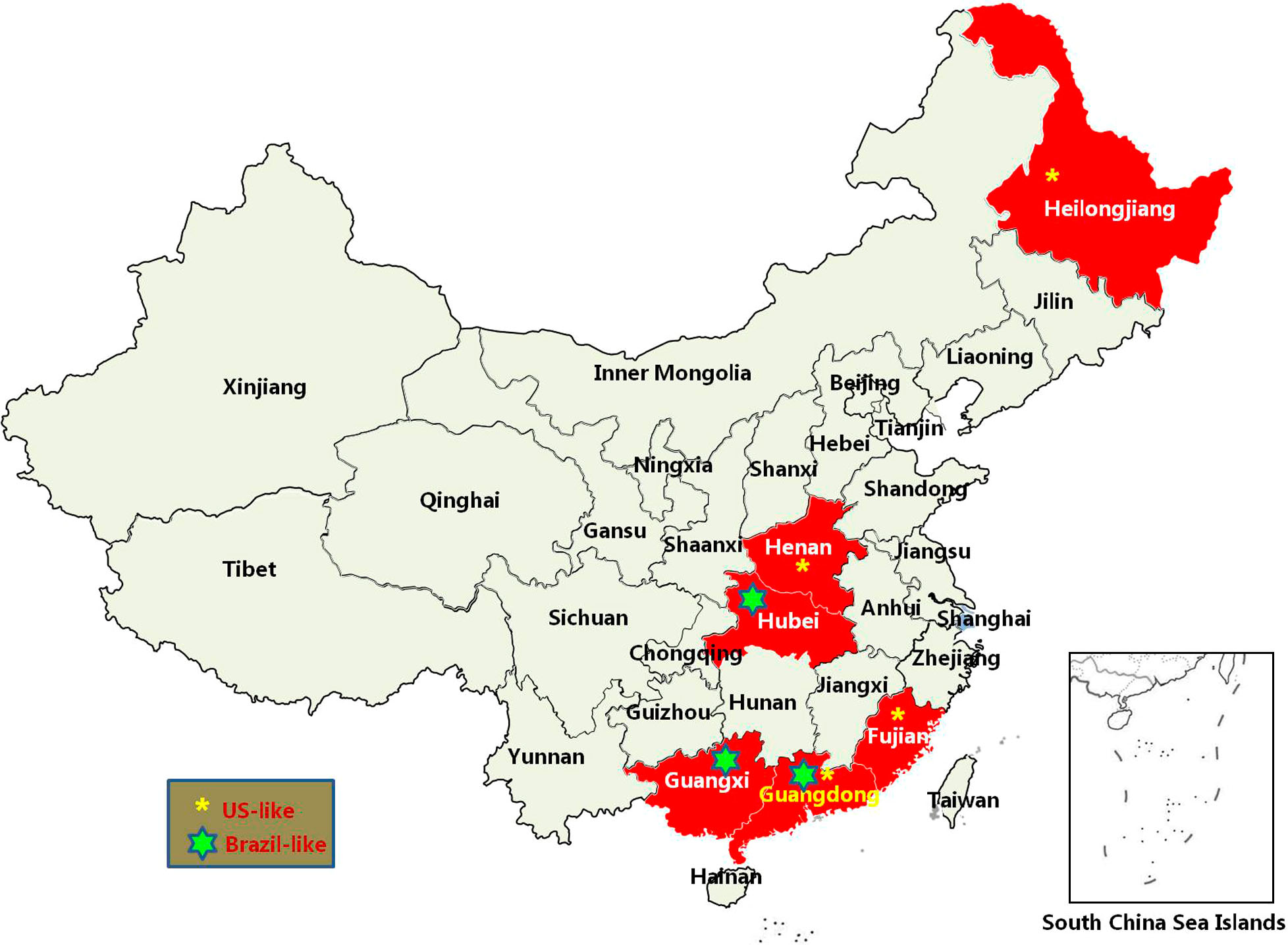
Figure 3. The current distribution of SVV in China. Red indicates the prevalence of SVV outbreaks.


SVV Isolation, Identification and Detection
Genomic Sequencing of the Two SVV Strains
Genetic Analysis of the Two SVV Strains
Phylogenetic Analyses
Amino Acid Mutations in CH-GD-2017-1 and CH-GD-2017-2, Compared to SVV-001 and SVV CH-01-2015
Global Geographical Distribution of SVVs
-
SVV, an emerging picornavirus of swine, causes vesicular disease and neonatal losses (Canning et al. 2016; Montiel et al. 2016). SVV has recently emerged in major swine producing countries around the world (Leme et al. 2017). An increased number of cases of SVV have been reported in the US and China (Joshi et al. 2016; Zhu et al. 2017). In China, SVV infection in pigs has been reported in Guangdong, Hubei, Fujian, Henan, Guangxi, and Heilongjiang provinces (Qian et al. 2016; Wu et al. 2016; Wang et al. 2017; Zhao et al. 2017; Zhu et al. 2017). The first SVV infection was reported in Guangdong Province in 2015 (Wu et al. 2016), and reemerged in 2016 (Zhao et al. 2017). This study identified new cases in Guangdong Province in 2017. We found that most SVV infections in China were reported in Guangdong Province, where farms often have large pig populations with high densities. The intensive feeding environment might have facilitated the spread of SVV and continuous outbreaks of SVV in pigs would seriously threaten the breeding industry. The US-like strains might have become the dominant epidemic strains within Guangdong in 2017. Currently, there are no well-established control strategies to limit the spread of SVV infection in pigs in China. Therefore, continuous epidemiology studies may play an important role for surveillance of the state of SVV in pigs.
As a picornavirus, SVV is RNA virus exhibiting higher rates of spontaneous mutation (Domingo and Holland 1997). The new SVV strains of CH-GD-2017-1 and CH-GD-2017-2 contained 28 mutated amino acids compared to the first Chinese SVV isolate, SVV CH-01-2015, and had 55 mutated amino acids compared to the prototype SVV isolate, SVV-001. This indicates that the persistent mutation of SVV and genetic diversity appeared over a short timeframe.
Amino acid substitutions may confer advantages that increase the fitness of the virus in pigs and result in the spread of the virus. The phylogenetic analysis suggested that all recent US strains were clustered in the same branch. The SVV strains from different countries were also located close to each other. The viral genome sequence was diversified between samples of different countries, resulting in distinct clusters observed in the phylogenetic trees. The SVV strains in China showed different features. Several strains were closely related to the US strains, while several strains were closely related to the Canadian or Brazilian strains. Those findings suggested the complicated spreading status of SVV in China (Fig. 3). The current Guangdong strains in 2017 also showed a distant relationship with previous Guangdong strains. The Chinese SVV strains isolated in 2017 were all clustered with the US-like strains, suggesting the US-like SVV strains were predominant in China in 2017.
The SVV has emerged in the major pig producing countries (Fig. 2). The reasons for the quick spread of SVV in different countries should be investigated. Transmission of SVV likely occurs via the oronasal route (Chen et al. 2016; Montiel et al. 2016; Maggioli et al. 2017). However, SVV has been detected from mouse feces, mouse small intestine and environmental samples. SVV RNA has also been detected from houseflies in pig farms (Joshi et al. 2016), suggesting that the pests in pig farms might also be a transmission route for SVV. In addition, both the domestic and international livestock and meat product trade might have promoted the spread of SVV in different regions and countries. SVV may have a wide range of transmission routes. However, whether these factors are extremely responsible for the rapid spread of SVV throughout the world remains unknown.
SVV mutates quickly and in ways that may affect current diagnostics and future vaccines. The large-scale occurrences of SVV infection will increase its mutation and the risk of spread. Therefore, additional attention should be paid to the mechanisms by which SVV spreads. Long-term surveillance programs and proper control policies should also be implemented in many countries. In conclusion, this study enriches our knowledge of the spread and geographical distribution of SVV in China in 2017. This study also provides valuable information for assessing the potential risk of SVV in pigs.
-
This work was supported by grants from the National Natural Sciences Foundation of China (No. U1501213), the Key Development and Research Foundation of Yunnan (2018BB004) and the Project Supported by National Science and Technology Ministry (2015BAD12B04).
-
PC, FY, ZZ and HZ designed the research, PC, FY, ZZ, WC and HL performed the experiments. KZ and XL provided experiment support. PC, ZZ and HZ wrote the manuscript. All authors have read and approved the final manuscript for submission.
-
The authors declare that they have no conflict of interest.
-
This article does not contain any studies with human or animal subjects performed by any of the authors.
Conflict of interest
Animal and Human Rights Statement
-

Table Supplementary Table S1. The primers used in this study.
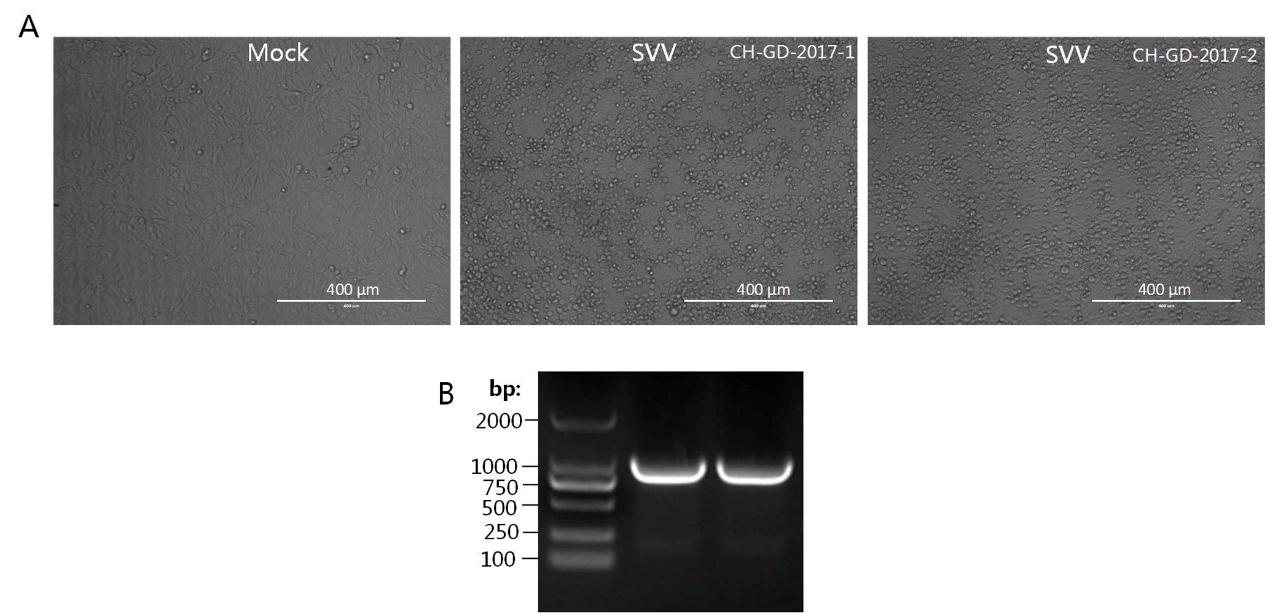
Figure Supplementary Figure S1. Identification of two SVV strains from Guangdong Province in 2017. A. BHK-21 cells were mock-infected or infected with CH-GD-2017-1 and SVV CH-GD-2017-2. The CPE was observed at 12 hours postinfection. B. Amplification of VP1 genes from CH-GD-2017-1 and SVV CH-GD-2017-2 isolated from the virus-infected BHK-21 cell cultures.
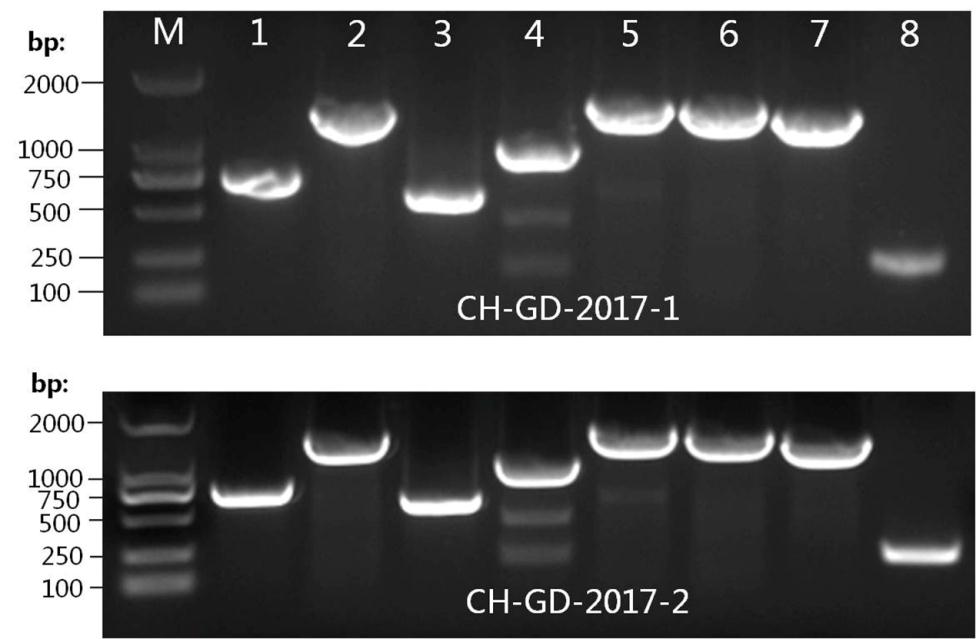
Figure Supplementary Figure S2. Amplification of different regions of viral genomes of CH-GD-2017-1 and SVV CH-GD-2017-2. M represented DNA ladder. Lanes 1¬8 represented the eight fragments amplified by the eight pairs of primers listed in Supplementary Table S1.

 DownLoad:
DownLoad: