














HTML
-
Japanese encephalitis virus (JEV) is a member of the genus Flavivirus within the family Flaviviridae. It is one of the most important mosquito-borne viruses and causes serious viral-encephalitis in East and Southeast Asia (Mackenzie et al. 2004; Sucharit et al. 1989; van den Hurk et al. 2009). Recently, this virus has spread to other geographic areas such as Australia, Pakistan, and Saipan following global warming and urbanization (Igarashi et al. 1994; Johansen et al. 2001; Mackenzie 2005; Mitchell et al. 1993). Japanese encephalitis (JE) has a mortality of 10%–50%, and approximately half of JE survivors have severe neurological sequelae (Turtle and Solomon 2018). Although vaccines against JEV have been used for many years, more than 50, 000 meningoencephalitis cases and approximately 15, 000 deaths are reported each year (Ginsburg et al. 2017; Turtle and Driver 2018).
The JEV genome is a single-stranded positive-sense RNA of approximately 11 kb. It encodes a single large polyprotein, which is cleaved by viral and host proteases to produce three structural proteins (capsid, premembrane, and envelope) and seven nonstructural (NS) proteins (NS1, NS2A/B, NS3, NS4A/B, and NS5) (Morita et al. 2015; Unni et al. 2011). NS1 protein is a nonstructural protein involved in many crucial biological events during viral infection and spreading, such as viral replication and virulence, immune invasion, and activation of the host complementary system (Amorim et al. 2014; Muller and Young 2013; Rastogi et al. 2016). NS1', a 52 amino acid C-terminal extension of NS1, was first detected in JEV-infected cells generated by a -1 programmed ribosomal frameshift (PRF) via a slippery heptanucleotide (YCCUUUU) and potential pseudoknot nearby (Blitvich et al. 1999; Firth and Atkins 2009). It has been reported that NS1' in West Nile virus (WNV) and JEV play important roles in neuroinvasiveness, but the mechanism has not been clarified (Melian et al. 2010; Wang et al. 2015; Ye et al. 2012).
As one of the most crucial host defense molecules, type I interferon (IFN-I) is essential for combating viral infections. To improve better survival, viruses develop different strategies to antagonize IFN-I (Snell and Brooks 2015; Teijaro 2016). Efficient replication of flaviviruses is closely linked with their ability to inhibit IFN-I production and signaling pathways (Best 2017; Urosevic 2003). For example, WNV NS1 inhibits IFN-I production by targeting RIG-I and MDA5 (Zhang et al. 2017); Dengue (DENV) and Zika virus NS5 antagonizes IFN-I signaling by degrading STAT2 (Ashour et al. 2009; Grant et al. 2016). However, few functions have been reported for flavivirus NS1' in inhibiting IFN-I production and signaling pathways.
In this study, a defective NS1' virus was generated to investigate the mechanism of how NS1' protein contributes to JEV pathogenesis. We found that the lower virulence of defective NS1' virus is related to IFN-I signaling in the host. Mechanistically, we demonstrated that NS1' protein antagonizes the production of IFN-β and IFN-stimulated genes (ISGs), promoting the replication of JEV.
-
Baby hamster kidney (BHK-21) cells, mouse testicular Sertoli (TM4) cells, African green monkey kidney (Vero) cells, human cervix epithelial (HeLa) cells, and human lung epithelial (A549) cells were stored in the laboratory and cultured in Dulbecco's Modified Eagle's Medium (DMEM; Sigma, St. Louis, MO, USA) supplemented with 10% fetal bovine serum (GIBCO, Grand Island, NY, USA), 100 U/mL penicillin, and 100 mg/mL streptomycin (Sigma) at 37℃ and 5% CO2.
The parental JEV (pSA14) and NS1' defective viruses (rG66A, a 'G'-to-'A' mutation at nucleotide position 66 in the NS2A gene of SA14 of JEV) were produced by electroporation of BHK-21 cells with transcribed RNA from the full-length cDNA clones pMW218-JEV-rAT and pMW218-JEV-NS2A-G66A, respectively, which were kindly provided by Fan Jia (Wuhan Institute of Physics and Mathematics, Chinese Academy of Sciences).
-
The plasmids pcDNA3.1-NS1 and pcDNA3.1-NS1' encoding the JEV NS1 protein and NS1' protein were constructed by polymerase chain reaction (PCR) using JEV-SA14 cDNA as a template and then cloned into the pcDNA3.1(+) vector. The primers were listed in Supplementary Table S1.
-
Mouse monoclonal antibody against JEV NS1 (2B8) and NS1' was generated in our laboratory. Briefly, mice were immunized with purified JEV NS1 protein and the monoclonal antibodies were produced and isolated by using hybridoma technology. Clone 2B8 which shows the highest specificity and sensitivity against JEV NS1 was used in this study. Commercial antibodies, including anti-GAPDH antibodies (ABclonal Technology, Woburn, MA USA), horseradish peroxidase-conjugated goat anti-mouse IgG (Boster, Wuhan, China), and Alexa Fluor 488-conjugated goat anti-mouse IgG (Invitrogen, Carlsbad, CA, USA) were used.
-
Cell lysates were generated in cell lysis buffer (Beyotime Biotechnology, Shanghai, China) for 20 min at 4 ℃. The protein concentration was determined by using a BCA protein assay kit (Thermo Scientific, Waltham, MA, USA) and boiled at 95 ℃ for 15 min. Equivalent amounts of proteins were separated by sodium dodecyl sulfate polyacrylamide gel electrophoresis (12% polyacrylamide) and electroblotted onto a polyvinylidene fluoride membrane (Millipore, Billerica, MA, USA) using a Mini Trans-Blot Cell (Bio-Rad, Hercules, CA, USA). The membrane was incubated with the relevant antibodies for 2 h at room temperature (RT, approximately 20–30℃) or overnight at 4℃. After washing three or four times with TBST (50 mmol/L Tris–HCl, 150 mmol/L NaCl, and 0.1% Tween 20, pH 7.4), the membrane was incubated with horseradish peroxidase-labeled goat anti-mouse IgG at RT for 30–45 min, and then visualized with a chemiluminescence system (Bio-Rad).
-
HeLa cells in 12-well plates (at a density of 2 × 105/well) were transfected with the relevant expression plasmids (pcDNA3.1-NS1 or pcDNA3.1-NS1') or an empty control plasmid (pcDNA3.1) (800 ng) using Lipofectamine 2000 (Invitrogen) for 24 h and then transfected with 5 μg poly(I:C) for another 12 h. For viral infection experiments, HeLa cells were transfected with plasmids for 12 h and then infected with JEV-pSA14 or rG66A for another 24 h.
-
Total RNA of cells was extracted according to the instructions of TRIzol Reagent (Invitrogen). The concentration of RNA was measured, and cDNA was synthesized by reverse transcription using a ReverTra Ace RT kit (Toyobo, Osaka, Japan). Real-time PCR was performed according to the instructions of the SYBR green real-time PCR master mix (Toyobo) and fluorescence intensity was analyzed with an ABI StepOne Plus system (Applied Biosystems, Foster City, CA, USA). The results were normalized to β-actin expression in each sample. The primers were listed in supplementary Table S1.
-
A549 or TM4 cells were infected with pSA14 and rG66A virus at a multiplicity of infection (MOI) of 1.0. At 12, 24, 36, and 48 h post-infection, the supernatant was harvested and stored at -80℃. The samples were used to infect BHK-21 cells by serially 10-fold dilution in 12-well plates. After incubation for 1.5 h, the cells were washed with PBS and cultured in DMEM containing 2% fetal bovine serum and 1.5% sodium carboxymethyl cellulose (Sigma) for 4 or 5 days. Next, visible plaques were counted, and viral titers were calculated.
-
Adult C57BL/6 J mice (female, 5-weeks-old, HZAUMO- 2017-016) were obtained from Scientific Ethic Committee of Huazhong Agriculture University (Wuhan, China), while IFNAR knockout (IFNAR-/-) C57BL/6 J mice were kindly provided by Bin Wei (Wuhan Institute of Virology, Chinese Academy of Sciences). The mice were maintained according to Committee for Protection, Supervision, and Control of Experiments on Animals guidelines, Huazhong Agricultural University.
Five-week-old C57BL/6 J mice were injected intracranially with 20 μL containing 200 PFU of pSA14 and rG66A virus diluted with DMEM. Control animals were injected with 20 μL serum-free DMEM by the same route. The day of injection was referred to as day 0. Five-weekold C57BL/6 J or IFNAR-/- mice were injected intraperitoneally with 200 μL containing 1 × 106 PFU of pSA14 and rG66A virus diluted with DMEM. Control animals were injected with 200 μL DMEM by the same route.
The presentation of clinical signs of disease and mortality in mice were monitored every day. Clinical signs of disease were divided into seven grades, including subclinical, ruffled fur and hunched, hindlimb weakness, mild paresis, moderate paresis, severe paresis, and moribund or dead (Daniels et al. 2017).
-
Statistical analyses of all data were conducted by using Prism (6.0) (GraphPad, Inc., San Diego, CA, USA). The log-rank test was used to analyze survival experiments. The differences in all other experiments were analyzed by Student's t test or two-way analysis of variance. A P value < 0.05 was considered as significant for all comparisons.
Cells and Viruses
Plasmid Construction
Protein and Antibodies
Western Blotting
Transfection
Quantitative RT-PCR
Plaque Assay
Inoculation of Mice with JEV
Quantification and Statistical Analysis
-
Nucleotide 66 of the NS2A gene is critical for producing the NS1' protein. The G 66 in the NS2A coding region was mutated into an A in pMW218-JEV-rAT, which is the fulllength cDNA clone of the JEV SA14 strain (Fig. 1A). This silent point mutation did not alter the amino acid sequence of NS2A protein. To clarify whether NS1' protein function was abolished, we infected BHK-21 cells with wild-type virus (pSA14) and NS1' defective virus (rG66A) at an MOI of 1, and cell lysates were harvested at 48 h post-infection. As expected, both NS1 and NS1' protein were detected in pSA14-infected BHK-21 cells, while only NS1 protein was detected in rG66A-infected BHK-21 cells (Fig. 1B).
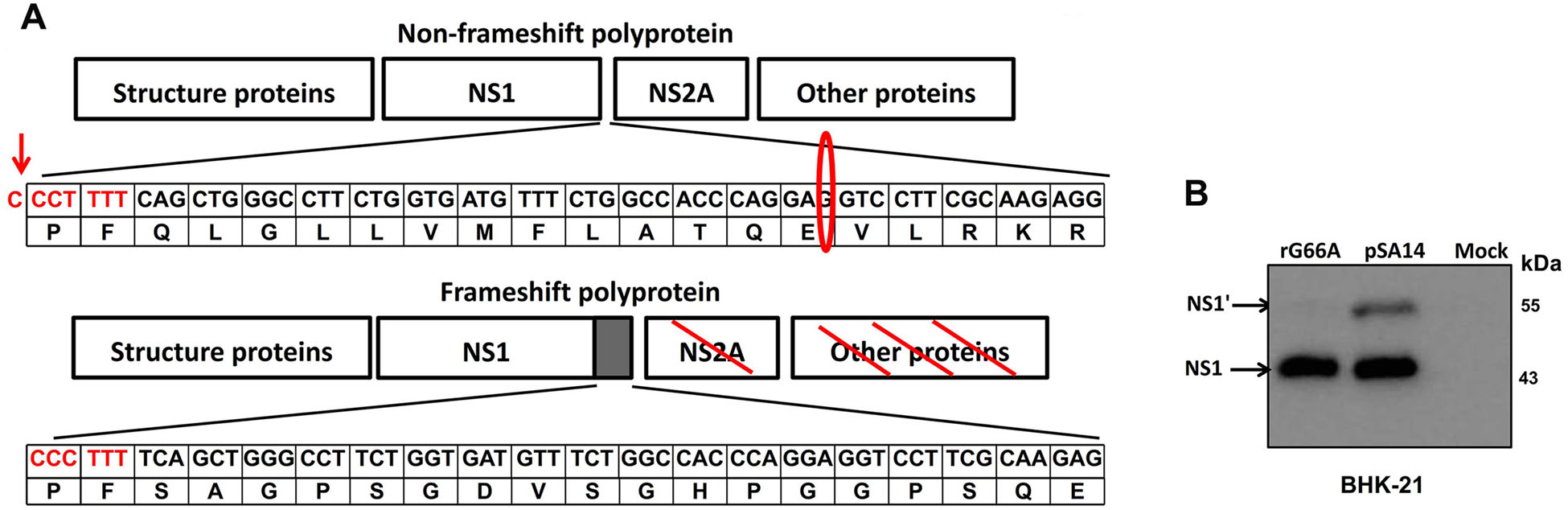
Figure 1. Recovery and characterization of the rG66A mutant virus. (A) Schematic depiction of the translational pathway of non-PRF and PRF flavivirus polyprotein. The sequence of flammulated letters indicates the slippery hepta-nucleotide frameshift motif and in flammulated oval is the silent nucleotide mutation (G → A) in pSA14 virus contributing downstream pseudoknot interactions. Flammulated arrow indicates where a -1 programmed ribosomal frameshift in pSA14 virus occurs. The frameshift polyprotein was aborted by a termination codon at 52 amino acids after C-terminal extension of NS1. Numbers in the frame represent nucleotides and amino acids in NS2A gene of JEV SA14. (B) BHK-21 cells were infected or mock-infected with pSA14 or rG66A viruses. NS1 and NS1' proteins were detected by western blotting with anti-NS1 monoclonal antibody (2B8) at 48 h post-infection
-
To determine the role of NS1' in JEV infection, we compared the neuro-virulence and peripheral-virulence of rG66A and WT virus in C57BL/6 J mice (5-week-old) by intracerebral (i.c.) or intraperitoneal (i.p.) injection. All mice injected intracerebrally with 200 PFU of pSA14 or rG66A virus died within 9 days (Fig. 2A). The survival and clinical signs of disease in mice infected with rG66A virus were indistinguishable from those of wide typeinfected controls. However, compared to pSA14 virus, both morbidity and mortality showed a slight delay in rG66Ainfected mice (Fig. 2A, 2B).
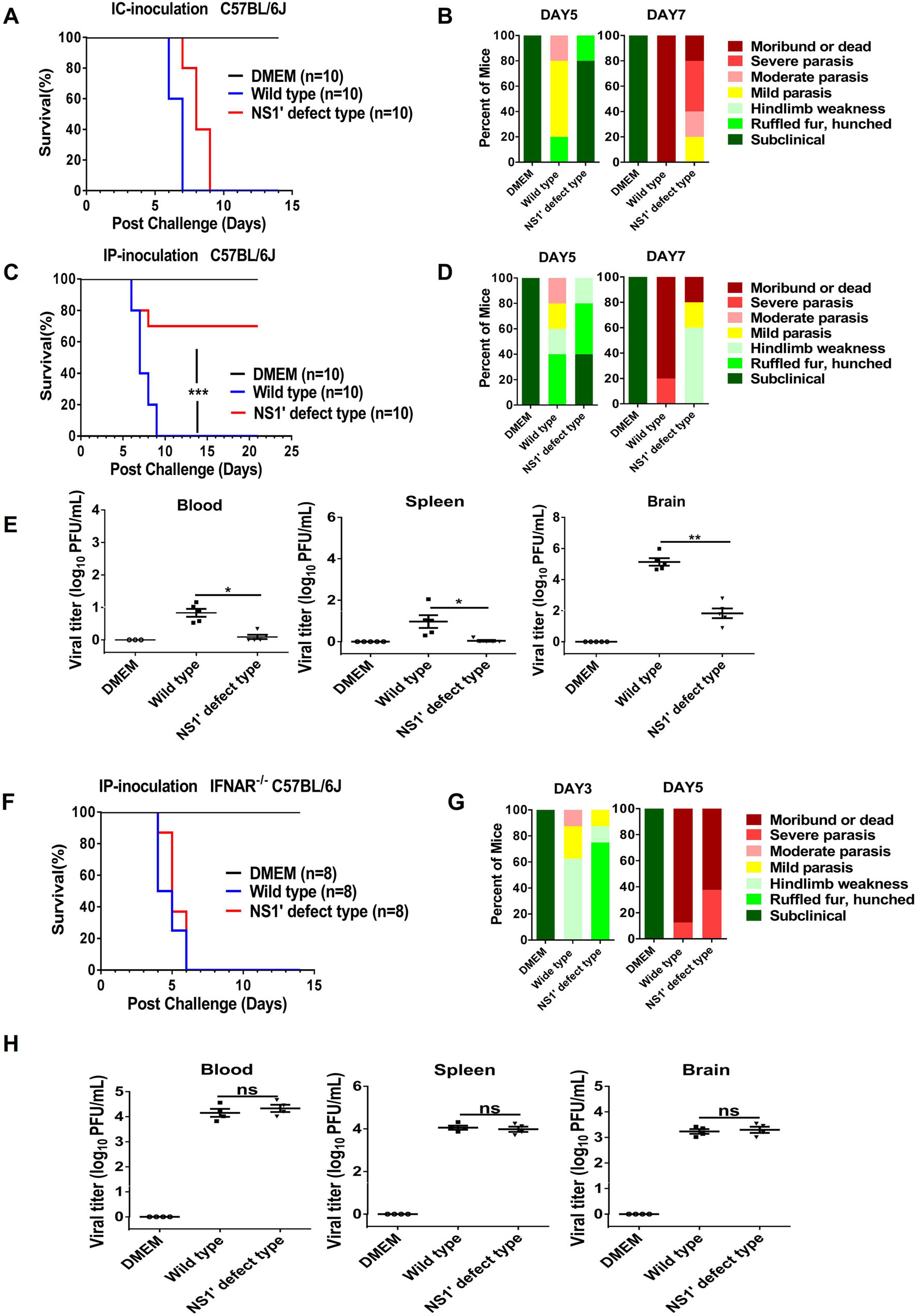
Figure 2. Comparison of neurovirulence of pSA14 and rG66A virus in mice. A, B Groups of 5-week-old C57BL/6 J mice (n = 10 per group) were infected with 2 × 102 PFU through the intracranial route. C, D Groups of 5-week-old C57BL/6 J mice (n = 10 per group) were infected with 1 × 106 PFU through the intraperitoneal route. E, G Groups of 5-week-old IFNAR-/- C57BL/6 J mice (n = 8 per group) were infected with 1 × 106 PFU through the intraperitoneal route. Survival curves are presented (A, C, and E). Presentation of clinical signs of disease on indicated days are presented (B, D, and G). Five-week-old C57BL/6 J (F) mice or IFNAR-/- C57BL/6 J (H) mice were infected intraperitoneally with 1 × 106 PFU of WTpSA14 and rG66A mutant virus. At day 5 following infection, the blood, spleen, and brain were harvested, weighed, homogenized, and assayed for JEV titers via plaque assay. *P < 0.05; **P < 0.01; ***P < 0.001. Error bars represent SEM. ns, no significance. All data are pooled from two independent experiments
Following i.p. injection, all mice infected with pSA14 virus died within 9 days after infection, while only 30% mortality were observed in mice infected with rG66A at day 9 post-infection (Fig. 2C). Higher mortality in pSA14- infected mice was accompanied by earlier and more severe development of clinical signs of disease (Fig. 2D). The typical neurological symptoms of rG66A-infected mice were significantly alleviated. Moreover, lower viral titers were detected in mouse the blood, spleen, and brain in rG66A group at 5 days post-infection (Fig. 2E). Taken together, these data indicate that NS1' protein plays an important role in viral multiplication in the peripheral tissue.
To clarify whether the different virulence levels of the two viruses in C57BL/6 J mice are linked to the IFN-I response, an IFNAR-/- mouse model (5-week-old) was used. As shown in Fig. 2F and 2G, all mice infected with pSA14 or rG66A virus died within 6 days after infection, and the clinical signs caused by rG66A virus were consistent with those caused by its parental virus. Similar viral loads were observed in the IFNAR-/- mouse blood, spleen, and brain at 5 days post-infection (Fig. 2H). These data collectively suggest that the difference between rG66A and pSA14 virus is linked to the inhibitory effect of NS1' on the IFN-I response.
-
Next, viral propagation of rG66A and pSA14 was measured in TM4 and A549 cells, which are susceptible to JEV. We found that growth of the rG66A mutant was somewhat reduced compared to pSA14 virus (Fig. 3A, 3B). However, after treatment with an antiIFNAR polyclonal antibody in the culture media of TM4 and A549 cells for 2 h before infection, the growth difference between the two viruses disappeared (Fig. 3C, 3D). To confirm this result, the growth properties of JEV rG66A and pSA14 were determined in Vero cells, which are IFN deficient cells. Similar replication efficiencies were observed between pSA14 and rG66A (Fig. 3E). These results suggest that reduced propagation of rG66A mutant is related to the IFN-I response. Next, the IFN-I response induced by JEV was analyzed. We found that rG66A virus induced higher IFN-β and ISGs such as ISG15, OAS-1, and MX-1 than that by pSA14 virus in TM4 cells and A549 cells (Fig. 3F, 3G). Collectively, these results suggest that compared to the wild-type virus, the rG66A mutant induces a greater IFN-I response, which may contribute to decreased viral propagation in host cells.
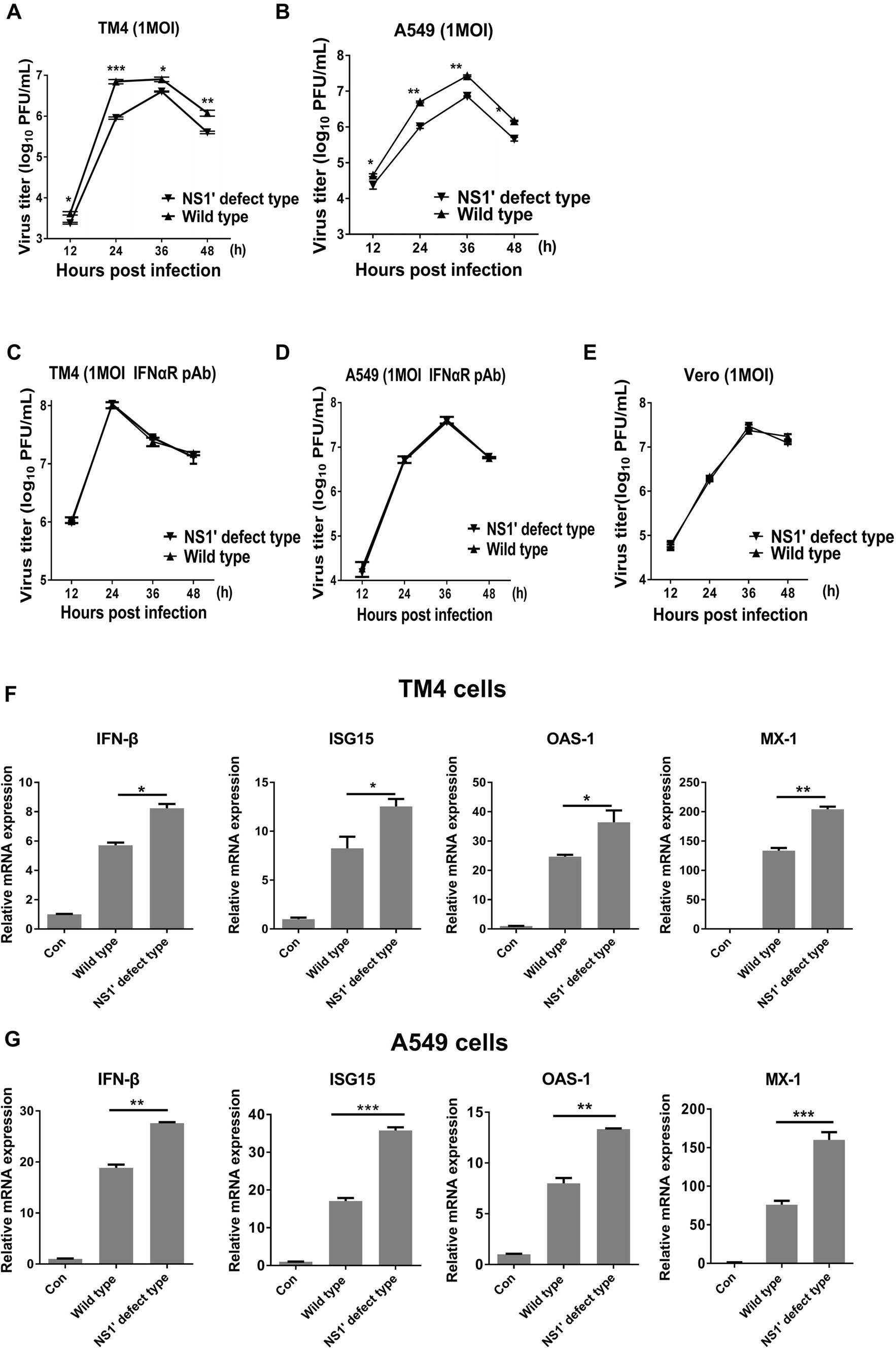
Figure 3. NS2A G66A mutation enhances induction of IFN-β and ISGs expression in cells. A, B Kinetics of replication of WT-pSA14 or rG66A mutant virus in TM4 (A) or A549 (B) cells. Cells were infected at an MOI of 1, and the viral titer was measured at 12, 24, 36, and 48 h post-infection using plaque assay of harvested cell supernatant. Growth kinetics from a typical experiment are shown. C, D Replication kinetics of WT-pSA14 or rG66A mutant virus in TM4 (C) or A549 (D) cells treated with IFNAR pAb. TM4 and A549 cells were treated with pAb followed by infection of pSA14 or rG66A at an MOI of 1. Viral titer was measured at 12, 24, 36, and 48 h postinfection using plaque assay with harvested cell supernatant. Growth kinetics from a typical experiment are shown. (E) Replication kinetics of WT-pSA14 and rG66A mutant virus in Vero cells. Cells were infected with pSA14 or rG66A at an MOI of 1, and the viral titer was measured at 12, 24, 36, and 48 h post-infection using plaque assay with harvested culture fluids. Growth kinetics from a typical experiment is shown. F, G TM4 (F) and A549 (G) cells were infected with WT-pSA14 or rG66A mutant virus (MOI = 1). mRNA levels of IFN-β, ISG15, OAS-1, and Mx-1 were determined by qRTPCR at 24 h post-infection. *P < 0.05, **P < 0.01, ***P < 0.001. All data are pooled from two or three independent experiments.The results were normalized to β-actin expression in each sample. Con means control
-
The above results suggest that NS1' plays a critical role in inhibiting IFN-I. To confirm this prediction, HeLa cells were transfected with plasmids encoding NS1 and NS1', and similar amounts of NS1 and NS1' protein were detected at 36 h post-transfection (Fig. 4A). The results showed that NS1' rather than NS1 suppressed poly(I:C)- triggered IFN-I production and response (Fig. 4B).
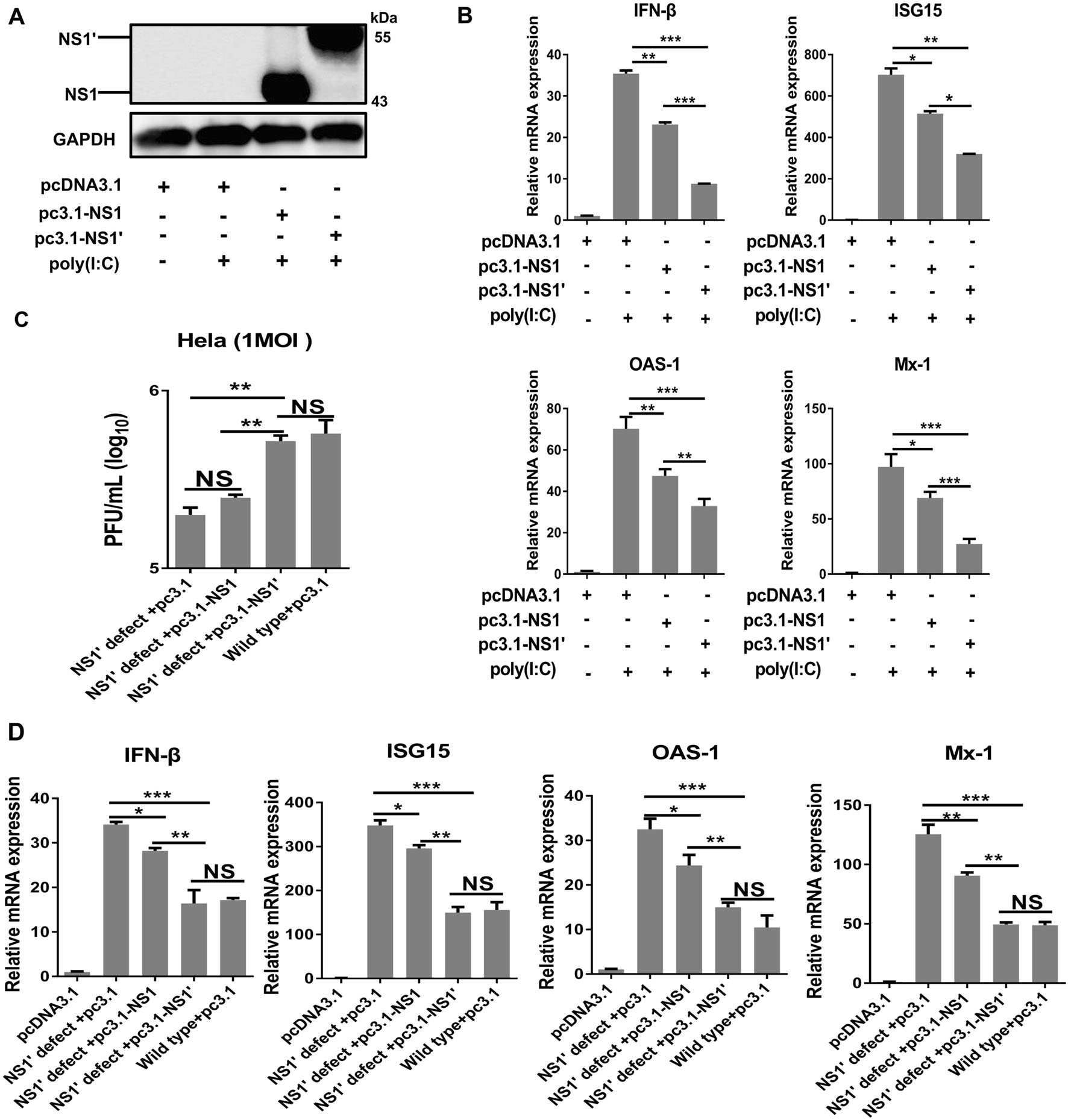
Figure 4. JEV NS1' reduces poly(I:C) triggered IFN-β and ISGs. A HeLa cells were transfected with empty vector (EV) or plasmid encoding NS1 or NS1'. Expression of JEV NS1/NS1' protein was determined by immunoblotting at 24 h post-transfection with an antiNS1 antibody (2B8), and GAPDH served as a reference control. B HeLa cells were transfected with an EV or plasmid encoding NS1 or NS1', followed by treatment with or without poly(I:C). After 24 h of incubation, the mRNA levels of IFN-β, ISG15, OAS-1, and Mx-1 were determined by qRT-PCR. C Kinetics of replication of WTpSA14 or rG66A mutant virus in HeLa cells. Cells were transfected with an empty vector or expression vector encoding NS1 or NS1' for 12 h and then infected with WT-pSA14 or rG66A mutant virus at an MOI of 1 for 24 h. Growth kinetics from a typical experiment are shown. D HeLa cells were transfected with EV, or plasmid encoding NS1 or NS1', followed by infection with WT-pSA14 or rG66A mutant virus at an MOI of 1. mRNA levels of IFN-β, ISG15, OAS-1, and Mx-1 were determined by qRT-PCR at 24 h post-infection. *P < 0.05, **P < 0.01, ***P < 0.001. ns, no significance. All data are pooled from two or three independent experiments. The results were normalized to β-actin expression in each sample
To further assess the effect of NS1' on inhibiting IFN-I during JEV replication, HeLa cells transfected with pcDNA-NS1, pcDNA-NS1', or pcDNA3.1 were infected with rG66A or pSA14. We found that transfection of NS1' led to increased growth of rG66A and reduced production of IFN-β and ISGs, which was similar to that of pSA14 (Fig. 4C, 4D). Taken together, these results demonstrate that NS1' protein antagonized the production of IFN-I and ISGs, promoting the replication of JEV.
G66A Mutation in JEV NS2A Abolishes NS1' Protein Production
NS2A G66A Mutation Reduces Virulence of JEV in Mice Relying on IFN-I Response
NS2A G66A Mutation Enhances IFN-β Expression in JEV-Infected Cells
JEV-NS1' Protein Reduces Poly(I:C)-Induced Expression of IFN-I
-
Compared to the well-known function of NS1, few studies of NS1' have been reported. A recent study showed that NS1' protein has the same function as NS1 during virus replication in cells, and animal experiments clearly demonstrated that NS1' protein enhances viral neuroinvasiveness (Young et al. 2013; Melian et al. 2010; Ye et al. 2012). Consistently, we showed that NS1'-deficient virus led to lower viral loads in the blood, spleen, and brain compared to its parental virus following i.p. injection. However, the two viruses showed the same virulence levels following i.c. inoculation. These findings suggest that NS1' protein plays a crucial role in viral propagation in the peripheral tissue, which may promote JEV entry into the central nervous system.
In this study, we found that JEV NS1' attenuated poly(I:C)-triggered IFN-I production. However, the molecular mechanism requires further analysis. Several studies clearly demonstrated the different approaches by which flaviviruses antagonize IFN-I production. For example, the NS4A protein of DENV sequesters MAVS to block IFN-I production, and WNV NS1 inhibits IFN-β responses by interacting with RIG-I and MDA5 (Dalrymple et al. 2015; Zhang et al. 2017). As observed for NS1 protein, NS1' protein has three forms, including a monomer (intracellular protein), membrane-bound protein (mNS1'), and secreted protein (sNS1') (Young et al. 2015). The region of NS1' that performs the IFN-I function will be explored in our next study.
Interestingly, NS1' is consistently produced by members of the JEV serogroup, such as JEV and WNV, which cause neuroinvasive disease, while other mosquito-borne flaviviruses such as DENV or YFV that produce nonneurological disease do not generate NS1', indicating that NS1' may play a role in neuroinvasion. Additionally, the transmission cycles of the JEV serogroup involve animals such as pigs and avian species, and studies are needed to determine whether NS1' plays a pivotal role in virus replication/transmission in the mosquito-bird/pig cycle.
In conclusion, we demonstrated that NS1' protein plays a crucial role in JEV pathogenesis. This is at least partially related to the inhibitory effect of JEV NS1' on the IFN-I response. Our findings provide insight into JEV immune evasion mechanisms and a novel potential target for developing therapeutic methods against JEV.
-
This work was supported by the National Key Research and Development Program of China (2016YFD0500407), National Natural Science Foundation of China (31502065 and 31572517), and Fundamental Research Funds for the Central Universities (2013PY051, 2662016Q003, and 2662015PY083).
-
JY, SBC, YFS, MC, and HCC conceived and designed the experiments. DYZ, FJ, QYL, LPZ, ZC, and ZKZ performed the experiments. DYZ analyzed the data. DYZ and JY wrote the manuscript and prepared the Figures. JY and SBC checked and finalized the manuscript. All authors read and approved the final manuscript.
Acknowledgements
Author Contributions
-
The authors declare no conflict of interests. All authors read and approved the final manuscript.
-
Animal experiments in this study were approved by the Scientific Ethics Committee of Huazhong Agricultural University (permit number HZAUMO-2017-016).
Conflict of interest
Animal and Human Rights Statement
-

Table Table S1. Primers used in this study

 DownLoad:
DownLoad: