











HTML
-
While the intracellular environment and embedded cellular machinery provide the needed vital force and necessary materials for viruses to replicate after infection, these host machineries are not available to these foreign invaders at ease. In fact, viruses have to counter the multiple layers of intracellular defense to replicate and establish their dominance for their propagation. RNA granules (Thomas et al.2011) are dynamic non-membrane subcellular structures (Ivanov et al.2018) containing translationally silenced messenger ribonucleoproteins (mRNPs), which play an important role in regulation of cellular homeostasis, RNA metabolism and gene expression at the posttranscriptional level (Anderson and Kedersha 2009). Stress granules (SG) and processing bodies (PB) (Eulalio et al.2007) are two of RNA granules well characterized in yeast and mammalian cells (Poblete-Duran et al.2016) and are important components of the host cell antiviral defense.
SG are non-membranous, transiently assembled cytoplasmic aggregates of 48S mRNPs and associated proteins (Stohr et al.2006; Buchan and Parker 2009), where stalled translation preinitiation complexes (PICs) repress the translation of nonessential mRNAs (Anderson et al.2015) and modulate cell signaling by sequestering key signal translation proteins (Kedersha et al.2013). Thus, SG are thought to be the aggregates of stable, translationally silent mRNAs (Kedersha and Anderson 2002). A variety of environmental stresses, including viral infection, can trigger SG formation in eukaryotic cells (Anderson and Kedersha 2008). In contrast, PB can exist in the absence of stress (Stoecklin and Kedersha 2013), which are sites of active mRNA decay (Decker and Parker 2012). SG initiate global translational arrest by storing mRNA (Anderson and Kedersha 2009) for exchange with either polysomes for translation or PB for degradation (Kedersha et al.2005). RNA-binding proteins TIA-1 (Kedersha et al.1999; Gilks et al.2004), G3BP (Tourriere et al.2003; Matsuki et al.2013) and PABP (Ma et al.2009; Smith and Gray 2010; Burgess et al.2011) are three fundamental components of SG during stress (Fig. 1). GW182 and de-capping/de-adenylating enzymes are specific components of PB (Kedersha et al.2005), where siRNA- or miRNA-guided mRNAs are processed and degraded (Liu et al.2005) (Fig. 1). Virus infection imposes stress on host cells (McInerney et al.2005) and thereby induces SG formation. SG can shut off the translation of bulk mRNAs (Poblete-Duran et al.2016) to regulate gene expression and compartmentalization of heterologous viral RNAs and proteins. At the same time, viruses must take strategies to confront these responses and maximize their own replication efficiency (White and Lloyd 2012) by inhibition of SG formation and disruption of PB assembly via virally encoded factors.
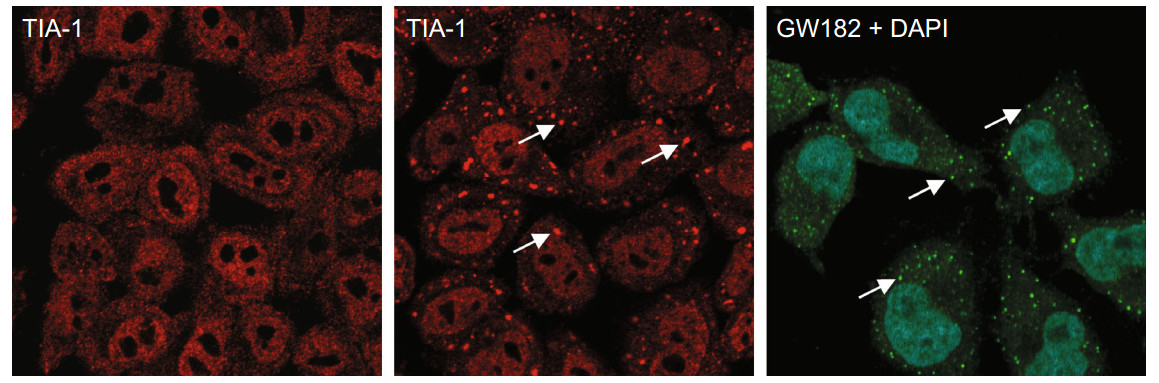
Figure 1. Mammalian RNA granules. HeLa cells immunostaining with anti-TIA-1 (left and middle, red) show stress granules (SG) during stress of NaAS2O3 (+arsenite, middle) and with anti-GW182 show processing bodies (PB) under physiological condition. Arrows indicate granules (SG or PB)
-
The process of SG formation can be artificially divided into the following steps (Fig. 2): (1) accumulation of stalled translation initiation complexes (Panas et al.2016) in response to various types of stress; (2) the RNA-binding proteins such as RAS-GTPase-activating protein SH3 domain-binding protein 1 (G3BP1) and T cell-restricted intracellular antigen 1 (TIA1) bind mRNAs and aggregate to nucleate SG formation. Self-aggregation of G3BP1 (Tourriere et al.2003) and the binding of TIA1 and TIAR (TIA-1-related protein) to polysome-free mRNAs, which exposes prion-like domains (Gilks et al. 2004), trigger mRNP aggregation. The aggregation of proteins is dynamic, and can rapidly exchange between SG and cytosol (Kedersha et al. 2000, 2005). (3) large SG aggregate from smaller foci via posttranslational modification and microtubule transport (McCormick and Khaperskyy 2017). Many SG proteins undergo multiple post-translational modifications (Jayabalan et al.2016; Protter and Parker 2016). For example, G3BP1 must be demethylated (Tsai et al.2016), dephosphorylated (Kedersha et al.2016) and poly(ADP)-ribosylated (Leung et al.2011) to promote SG nucleation. Accordingly, SG formation also requires ongoing transport of mRNPs along with an intact microtubule cytoskeleton (Ivanov et al.2003). Theoretically, viral interference with any of these important steps may modulate SG formation in cells. In fact, many viral factors can interfere with SG formation and/or function. Meanwhile, SG can entrap viral RNA in some cases (McCormick and Khaperskyy 2017). Therefore, SG are thought to be antiviral (Rozelle et al.2014). Thus, to illustrate the relationship between SG and RNA viruses would be important for us to better understand the interactions of host and viruses.
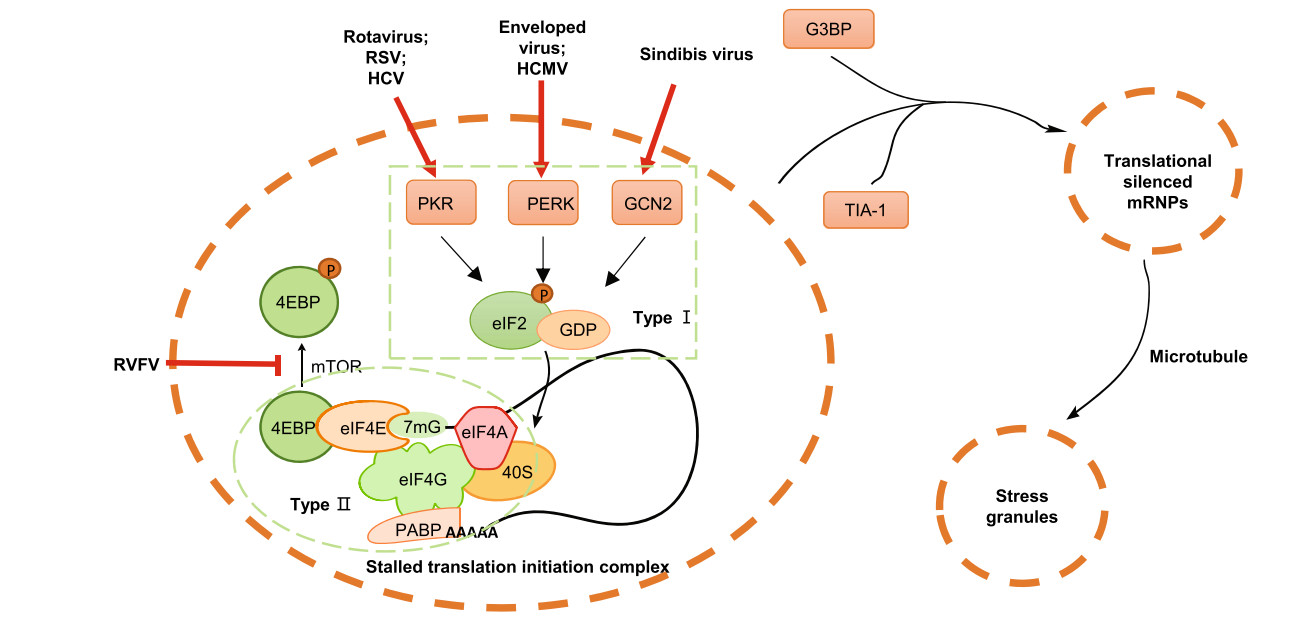
Figure 2. Viruses induce SG formation. Type Ⅰ SG formation: RNAs derived from rotavirus, RSV and HCV activate PKR; High levels of glycoproteins produced from enveloped virus activate PERK; HCMV infection activates PERK; Sindbis virus genomic RNA activates GCN2. Type Ⅱ SG formation: RVFV attenuates mTOR signaling to inhibite 4EBP phosphorylation. All above lead to the formation of stalled translation complexes to initiate the assembly of SG
Up to the present, SG can be divided into two types according to their formation mode. Type Ⅰ SG formation depends on phosphorylation of eukaryotic translation initiation factor-2α (eIF2α) by one of the eIF2 kinases—double-stranded RNA (dsRNA)-activated protein kinase or protein kinase R (PKR) (Srivastava et al.1998; Garcia et al.2007; Onomoto et al.2012), PKR-like ER kinase (PERK) (Harding et al.2000a, b), general control non-derepressible protein 2 (GCN2) (Wek et al.1995; Deng et al.2002) or haeme-regulated inhibitor (HRI) (McEwen et al.2005), which are activated by distinct types of stress. Phosphorylated eIF2α stably binds to eIF2β, which prevents the recycle of eIF2 and regeneration of the eIF2-GTP-Met-tRNAiMet ternary complex. Thus, eIF2α phosphorylation blocks recognition of the initiation codon and joining of the large ribosomal subunit, resulting in accumulation of stalled 48S mRNPs (Jackson et al.2010). Type Ⅱ SG formation is independent of eIF2α phosphorylation, but requires eIF4F complex disruption such as inhibition of eIF4A RNA helicase (Bordeleau et al.2006; Dang et al.2006) or disruption of eIF4E activity (von der Haar et al.2004; Fournier et al.2013) for recognition and binding of RNA cap structure. The stress induced by nutrient, energy, oxygen or growth factor insufficiency inhibits mTOR complex 1 (mTORC1), whose activity is required for the dissociation of 4EBPs from eIF4E (Fujimura et al.2012) and enables eIF4E to form the eIF4F complex, and thus blocks assembly of pre-initiation complexes (Zoncu et al.2011).
Type Ⅰ SG formation induced by viruses is the most and best-studied example (Table 1, Fig. 3A). Various RNA products derived by viruses including long dsRNA (Rojas et al.2010), 5′-triphosphate RNA (5′-ppp-RNA) (Nallagatla et al.2007), dsRNA that is formed by the antiparallel mRNA transcripts of some DNA viruses (Willis et al.2011), and human immunodeficiency virus (HIV) transactivation-response region (TAR) RNA hairpins (Heinicke et al.2009), can be recognized by PKR. The activated PKR initiates SG assembly through eIF2α phosphorylation. For instance, the persistent phosphorylation of eIF2α (Montero et al.2008) during rotavirus infection is PKR-dependent as a consequence of the accumulation of viral dsRNA in the cytoplasm outside the viroplasms (virus-induced cytoplasmic inclusion bodies called viroplasms [VMs]) (Rojas et al.2010). Even though eIF2α is phosphorylated in rotavirus-infected cells, the formation of SG is prevented and viral proteins are efficiently translated, suggesting that the virus prevents the assembly of these structures presumably downstream of eIF2α phosphorylation to allow the translation of its mRNAs (Mazroui et al.2006). Very recently, Dhillon and Rao found that rotavirus induces formation and sequestration of remodeled SG and PB in the VMs which contain the majorities of their components but selective exclusion of a few proteins (G3BP1 and ZBP1 for SG, DDX6, EDC4 and Pan3 for PB), to promote virus replication (Dhillon and Rao 2018). Oceguera et al. demonstrated that viral RNA of rotavirus could interact with several RNA binding proteins (RBPs) (Xrn1, Dcp1, Ago2, Hur) and interfere with their subcellular localization (Oceguera et al.2018). Lindquist et al. (Lindquist et al.2011) first determined that SG induction by respiratory syncytial virus (RSV) was mediated by PKR-dependent eIF2α phosphorylation. The RSV-mediated SG formation was significantly reduced in PKR-knockdown cells (Lindquist et al.2010). In addition, it has been shown that Hepatitis C virus (HCV) strongly activates PKR via the 5′-untranslated region (UTR) of its genome (Toroney et al.2010), thereby inducing SG. NS1-mutant Influenza virus A (IAV) (Khaperskyy et al.2012; Mok et al.2012; Ng et al.2013) and C protein-deficient Sendai virus (SeV) (Takeuchi et al.2008) lead to significant activation of PKR and eIF2α phosphorylation. Besides, PERK could be activated by high levels of glycoproteins produced from enveloped viruses (Chan and Egan 2005), and general control non-derepressible-2 (GCN2) could be activated by Sindbis virus (SINV) genomic RNA (Berlanga et al.2006), both leading to phosphorylation of eIF2α. GCN2 prevents replication of SINV in the early stages of the viral replicative cycle by blocking the synthesis of NSPs from SINV RNA (Berlanga et al.2006; Frolova et al.2006; Gorchakov et al.2008).

Table 1. Regulation of SG by viruses

Figure 3. Viruses interfer with SG formation. A Viruses modulate eIF2a phosphorylation. IBs of HPIV3 shield viral RNAs from recognition by PKR; IAV NS1, MERS-CoV accessory protein 4a, EBOV VP35, SeV and MV C protein and KSHV ORF57 prevent viral dsRNA from binding by PKR; ORF57 interacts with PACT to prevent PKR activation; HCMV pTRS1 and pIRS1 and HSV-1 vhs and Us11 block PKR activation; HCV NS5A and JEV NS2A interact with PKR and prevent PKR dimerization; N and GPC of JUNV impair the phosphorylation of eIF2a; HCV modulates GADD34 and PP1 to de-phosphorylate eIF2a. B Viruses modulate SG formation downstream of eIF2a phosphorylation. 3C protease of PV, EMCV, FMDV and CVB3 cleaves G3BP at Q326; FCV NS6 protein cleaves G3BP at E405; 2A protease of EV71, PV and CVA cleaves eIF4G at G689; L protein of both TMEV and mengovirus inhibits G3BP1 aggregation; DENV 30-UTR interacts with G3BP; SeV Trailer RNA captures TIAR from SG; WNV and Dengue virus (DENV) 30-end genome captures TIA-1/TIAR; HSV-2 vhs localizes to SG and its endoribonuclease activity is required to disrupt SG formation; EBOV and RSV sequester SG proteins within viral inclusion bodies; VV sequesters crucial SG components within DNA factories.
Viruses also induce SG formation independent of eIF2α phosphorylation (Table 1). The most typical example is from Rift Valley fever virus (RVFV) (Habjan et al.2009; Ikegami et al.2009) (Fig. 2). RVFV (Hopkins et al.2015) infection attenuates Akt/mTOR signaling and inhibits 4EBP phosphorylation and translation of 5′-TOP mRNAs, subsequently leading to an inhibition of global protein translation. 5′-TOP-containing mRNAs are indeed targeted to PB, where RVFV uses these cellular mRNAs for cap-snatching (Hopkins et al.2015). This can reflect that SG may interact with PB in a process that is thought to result in the exchange of mRNA cargos (Kedersha et al.2008). Whether any virus induces SG formation to cause translation inhibition due to the destruction of eIF4G or eIF4A is worth exploring in the future.
-
SG formation shuts off bulk host protein synthesis. However, all viruses depend on the host translation apparatus for their gene expression. Therefore, viruses, as intracellular parasites, have to modulate the stress response pathway and SG assembly to translate their proteins for virus replication. RNA viruses modulate stress response pathway at different levels of SG formation (Table 1): One is to regulate eIF2α phosphorylation, and the other is to regulate the process of SG nucleation.
-
In some cases, viral gene products can act as antagonists by targeting the virus-activated eIF2α kinases such as PKR or even by directly modulating the phosphorylation of eIF2α (Fig. 3A). IAV NS1 (Khaperskyy et al.2012; Ng et al.2013), Middle East respiratory syndrome coronavirus (MERS-CoV) accessory protein 4a (Rabouw et al.2016; Nakagawa et al.2018), and Ebola virus (EBOV) multifunctional protein VP35 (Nelson et al.2016; Le Sage et al.2017) bind viral dsRNA and prevent the viral dsRNA from PKR binding to inhibit SG formation. Inhibition of SG formation facilitates the translation of viral mRNAs, leading to efficient virus replication. HCV NS5A protein (Toroney et al.2010) binds to the PKR dimerization domain to inhibit PKR activation. Japanese encephalitis virus (JEV) NS2A protein (Tu et al.2012) might similarly interact with PKR and then prevent PKR dimer formation. SeV (Takeuchi et al.2008) and measles virus (MV) (Okonski and Samuel 2013) encode a C protein to limit the accumulation of dsRNA to inhibit SG formation. It seems that a portion of RNA viruses encode RNA binding proteins to antagonize the activity of PKR. There are also other groups of RNA viruses which directly modulate the phosphorylation of eIF2α without PKR. Junín virus (JUNV) prevents SG assembly by impairing the phosphorylation of eIF2α through its nucleoprotein (N) and glycoprotein precursor (GPC) (Linero et al.2011). However, its mechanism remains to be elucidated, although it may be similar to HCV. Ruggieri and colleagues reported that HCV rapidly de-phosphorylated eIF2α through protein phosphatase 1 (PP1) and its regulatory subunit GADD34 (growth arrest and DNA-damage-inducible 34) (Kojima et al.2003; Clavarino et al.2012; Ruggieri et al.2012).
-
Several RNA viruses have been shown to express viral effectors that can actively disrupt the accumulation of SG through cleavage of SG components (Fig. 3B). Poliovirus (PV) induces SG formation in early phase but induces SG disassembly at later stages via cleavage of G3BP by viral 3C, thus preventing SG formation (White et al.2007). Similar findings were also reported for encephalomyocarditis virus (EMCV) (Ng et al.2013), foot-and-mouth disease virus (FMDV) (Ye et al.2018; Visser et al.2019), coxsackievirus B3 (CBV3) (Fung et al.2013) and feline calicivirus (FCV) (Humoud et al.2016). FCV infection does not cause accumulation of SG, despite an increased phosphorylation of eIF2α (Humoud et al.2016). This is because FCV NS6Pro, a 3C-like proteinase, cleaves G3BP1 at a site different from the poliovirus 3C proteinase. Unlike FCV, murine norovirus (MNV) does not cleave G3BP1 and thus does not inhibit SG formation during virus infection (Humoud et al.2016). In general, picornaviruses inhibit SG formation by viral 2A/L or 3C cleaving the major components of SG. In recent study, Yang et al. found that the 2A protease of picornavirus (EV71, PV, CVA) inhibits typical SG formation, which is PKR and eIF2α phosphorylation-dependent, but induces atypical SG formation by cleaving eIF4GI to sequester cellular mRNA and release viral mRNA, thereby facilitating viral infection (Yang et al.2018). In other words, the 2A protease can transform the overall translation machinery favorable for productive viral infection by induction of atypical SG while blocking the typical SG in the presence of G3BP cleavage by viral 3C protease during viral infection (Yang et al.2018).
Redistribution or sequestering SG components to the viral replication sites is another strategy used by many viruses to impair SG assembly in infected cells (Fig. 3B). ZIKV infection induces the redistribution of TIAR to the viral RNA replication sites (Hou et al.2017); SeV Trailer RNA captures TIAR from SG (Iseni et al.2002); West Nile Virus (WNV) and Dengue virus (DENV) 3′-end viral genome captures TIA-1/TIAR (Li et al.2002; Emara and Brinton 2007; Xia et al.2015); DENV 3′-UTR interacts with G3BP1, G3BP2, Caprin1 and USP10 (Ward et al.2011; Reineke et al.2015); JEV recruits G3BP and USP10 to the perinuclear region through the interaction of JEV core protein with Caprin-1, a SG-associated cellular factor (Ward et al.2011). Theiler murine encephalomyelitis virus (TMEV) and mengovirus, a strain of EMCV, express the leader (L) protein to inhibit G3BP1 aggregation (Borghese and Michiels 2011). Sequestration or redistribution of SG components by viruses through protein-protein and protein-RNA interactions not only prevents SG assembly, but also facilitates viral genome replication. HCV-JFH1 infection redistributes several SG components, including G3BP1, ataxin-2 (ATX2), and poly(A)-binding protein 1 (PABP1), to the HCV replication complex (RC) (Ariumi et al.2011; Pene et al.2015), and co-opts G3BP1 to mediate efficient viral replication by interaction with NS5B and the 5′ end of the HCV minus-strand RNA (Ariumi et al.2011; Garaigorta et al.2012).
-
Studies on Human parainfluenza virus type 3 (HPIV3) (Hu et al.2018), RSV (Rincheval et al.2017), EBOV (Hoenen et al.2012), Rabies virus (RABV) (Lahaye et al.2009) and Vesicular stomatitis virus (VSV) (Heinrich et al.2010) showed that inclusion bodies (IBs) of negative stranded RNA viruses are the sites of viral RNA synthesis. A recent study suggested an emerging role of IBs in HPIV3 replication by shielding newly synthesized viral RNA from the antiviral effect of SG (Hu et al.2018) (Fig. 3B). Sequestration of O-linked N-acetylglucosamine (OGN) transferase (OGT), an enzyme that catalyzes the posttranslational addition of OGN to protein targets, in RSV IBs was also proposed to regulate SG nucleation and suppression of SG formation (Fricke et al.2013) (Fig. 3B). Viral transcription and replication of RABV take place within Negri bodies (NBs), which are IB-like structures (Lahaye et al.2009). RABV-induced SG are normally located closely to NBs. Viral mRNAs rather than viral genomic RNA accumulate in the SG-like structures together with cellular mRNAs were found to be specially transported from NBs to SG-like structures (Nikolic et al.2016). VSV infection also induces formation of the SG-like structures that co-localize with viral replication proteins and RNA, which are different from canonical SG (Dinh et al.2013). SG proteins (eIF4G, eIF3, PABP) are selectively sequestered within Ebola virus inclusion bodies and co-localize with viral RNA to form inclusion body-bound granules, which are functionally and structurally different from canonical SG, probably leading to inhibit the antiviral role of SG (Nelson et al.2016) (Fig. 3B). Collectively, these findings provoke more investigations on the roles of viral IBs in viral replication and resisting cellular responses.
-
Unlike RNA viruses, the regulation of SG formation during infection with DNA viruses is poorly understood. It was reported that human cytomegalovirus (HCMV) infection modifies the unfolded protein response (UPR) and activates PERK (Fig. 2), but limiting the amount of phosphorylated eIF2α to maintain translation (Isler et al.2005). Kaposi's sarcoma-associated herpesvirus (KSHV) ORF57 (Sharma et al.2017) interacts with PKR and PKR-activating protein (PACT) (Patel et al.2000) to inhibit PKR binding dsRNA and prevent PACT-PKR interaction in the PKR pathway (Li et al.2006), respectively. HCMV pTRS1 and pIRS1 antagonize PKR to facilitate virus replication (Ziehr et al.2016). The HSV-1 vhs (Sciortino et al.2013) and Us11 protein (Cassady and Gross 2002) play a key role in blocking the activation of PKR. Smiley and colleagues also demonstrated that infection with virion host shutoff protein (vhs)-defective herpes simplex virus 1 (HSV-1) triggers SG formation, and PKR is essential for SG formation in the absence of vhs (Dauber et al.2016) (Fig. 3A). Finnen et al. previously established that herpes simplex virus 2 (HSV-2) infection impacts stress granule accumulation in response to oxidative stress (Finnen et al.2012). They also demonstrated that disruption of SG is mediated by vhs (Finnen et al.2014), whose endoribonuclease activity is required to disrupt SG formation (Finnen et al.2016). HSV-2 vhs indeed have the ability to localize to SG (Finnen et al.2016) (Fig. 3B). This implies that removal of RNA from SG promotes its disassembly and that intact RNA is crucial for maintaining SG structure. It will be interesting to test the function of endoribonucleases in SG disassembly. Vaccinia virus (VV) sequesters crucial translation initiation factors, such as G3BP1, Caprin1, eIF4E, PABP and eIF4G (Katsafanas and Moss 2007; Simpson-Holley et al.2011; Zaborowska et al.2012), within cytoplasmic viral DNA factories to utilize SG components for different purposes (Fig. 3B). A recent study (Meng and Xiang 2019) suggested that the RNA granules are resulted from untranslated mRNA accumulation in viral DNA factories (Liu and Moss 2016) and TIA-1 is probably not required for granule formation and anti-poxviruses. Instead, the granules formation is most likely driven by an array of RNA-protein interactions and requires no specific SG components (Sivan et al.2018; Meng and Xiang 2019).
SG Formation and Induction of SG by RNA Virus Infections
RNA Viruses Modulate SG Formation or Assembly
RNA Viruses Modulate eIF2α Phosphorylation to Interfere with SG Formation
RNA Viruses Cleave/Sequester/Redistribute Stress Granule-Nucleating Proteins to Interfere with SG Assembly
RNA Virus Inclusion Bodies (IBs) Emerging as a New Strategy Used by Viruses to Resist SG
DNA Viruses Regulate SG Formation
-
PB were first reported in the scientific literature by Bashkirov et al.1997, and described as "small granules or discrete, prominent foci" or as the cytoplasmic location of the mouse exoribonuclease mXrn1p (Bashkirov et al.1997). Like SG, PB lack outer lipid membrane and now are recognized to be the sites where non-translating mRNAs accumulate for different fates including decay, storage, or returning to translation. A variety of enzymes involved in mRNA deadenylation (Ccr1, Caf1, Not1) (Sheth and Parker 2006), decapping (Dcp1/2, Lsm1-7, Edc3proteins) (Ingelfinger et al.2002; Yu et al.2005), nonsense-mediated decay (NMD) proteins (SMG5-6-7, UPF1) (Ingelfinger et al.2002; Durand et al.2007), in addition to scaffolding proteins (Ge-1/Hedls) (Yu et al.2005) and translation control factors (CPEB, eIF4E-T) (Andrei et al.2005; Wilczynska et al.2005), are the components of PB and used as routine markers to distinguish these granules. Nonetheless, some components (APOBEC3G, BRF1, DDX3, FAST, TTP, Rap55) (McEwen et al.2005; Sen and Blau 2005; Gallois-Montbrun et al.2007; Chen et al.2008) have also been shown to be shared by both SG and PB, suggesting a substantial linkage of these two structures and movement of mRNAs between both RNA granules. Interestingly, among these components, PB also include RNA-induced silencing complex (RISC) or miRNA associated argonaute (Ago) proteins (also shared with SG) and the GW182 protein which provides scaffolding activities for RISC to function, suggesting PB being the sites of miRNA mediated translation repression. The scaffolding activity of GW182 is critical for PB and knockdown of GW182 expression disrupts PB formation (Liu et al.2005). Notably, GW182 has been shown to bind to Ago2 which is critical for miRNA function and PB formation (Liu et al.2005). Recent evidence indicates that GW182 can recruit up to three molecules of Ago2 via its three GW motifs (glycine-tryptophan repeats) while each Ago protein has a single GW182-binding site (Elkayam et al.2017) (Fig. 4).

Figure 4. Disruption of PB assembly by viruses. The mRNA translation can be stopped for various reasons including the binding of miRNA. The translating mRNA can be stripped of ribosomes and the initiation complex can be collaps when binding to miRNA-RISC complex. The mRNPs targeted by PB components undergo three outcomes: 1. Translational inhibition; 2. Pan2/3-mediated deadenylation; 3. RNA decay by other associated RNA decay factors (e.g., Xrn1, Dcp1a, DDX6, and Lsm). Several RNA and DNA viruses which inhibit PB assembly are shown
By applying fluorescence-activated particle sorting to purify PB in combination with mass spectrometry, Hubstenberger et al. identified 125 proteins that are significantly associated with PB (Hubstenberger et al.2017). By labeling several PB-localized proteins with a BirA (E. coli biotin ligase) enzyme in combination with mass spectrometry after streptavidin pulldown, Youn et al. identified 38 proteins in the PB (Youn et al.2018). ISGs (interferon stimulated genes) can also be found in PB during virus infection (Hebner et al.2006).
-
In comparison to viral regulation of SG, interaction of virus and PB was not much explored. It is an assumption that RNA viruses must regulate RNA decay processes/machinery to prevent degradation of virus genomes and mRNAs. Recently, some progress has been made to understand the relationship between PB components and some viruses in the context of viral gene expression. The data in published literatures are summarized in (Table 2). Mutation induced in the PB core components to affect the viral life cycles are well studied and tabulated in an earlier review (Beckham et al.2007). The report linking the assembly of yeast Ty3 retrotransposons virus—like particles with PB presented the first link between human retrovirus and PB (Checkley et al.2010). The later study revealed PB to be the site of anti-viral host factors APOBEC3G and APOBEC3F (A3G or A3F, apolipoprotein B mRNA-editing enzyme catalytic polypeptide 1-like) family of cytidine de-aminases, presumably representing a component of innate immunity against HIV (Wichroski et al.2006; Gallois-Montbrun et al.2007). In a different study, A3F was found to specifically interact with cellular signal recognition particle RNA (7SL RNA). Efficient packaging of 7SL RNA and A3F into HIV virons was mediated by the RNA-binding nucleocapsid domain of HIV-1 Gag (Wang et al.2007).

Table 2. Regulation of PB assembly by viruses
The bona fide and unique dependence of viruses on PB came from the studies on plant brome mosaic virus (BMV) (Beckham et al.2007). This study suggested the accumulation of BMV mRNAs in PB was an important step in RNA replication complex assembly for BMV, and possibly for other positive-strand RNA viruses. Nonetheless, many RNA viruses initiate the process of transcription of viral RNA by the process of 'cap snatching' which involves the acquisition of capped 5′ oligonucleotides from cellular mRNAs. Interestingly, PB were shown to serve as a pool of primers in the case of Hantavirus while its nucleocapsid protein, which accumulates in PB, binds 5′ caps with high affinity (Mir et al.2008).
The base-pair complementarity between a miRNA and a target mRNA dictates the miRNA to specifically repress posttranscriptional expression of mRNAs. Subsequent events in this process involve relocation of RNA-induced silencing complexes (RISCs) together with several other RNA binding proteins to form PB. In this context, HIV-1 mRNA interacts with RISC proteins and disrupting PB structures enhances viral production and infectivity, suggesting a role of PB against viral infection (Nathans et al.2009). Specific miR-29a-HIV-1 mRNA interaction was found to enhance viral mRNA association with RISC and PB proteins and regulate HIV-1 production and infectivity. HIV Nef interacts with Ago2 via its glycine-tryptophan region and functions as a viral suppressor of RNAi (Aqil et al.2013). While overexpression of Mov10, a component of PB and an ATP-dependent 5′-3′ RNA helicase, inhibits HIV production (Burdick et al.2010; Furtak et al.2010), Mov10 and APOBEC3G localization to PB is not required for HIV virion incorporation and antiviral activity (Izumi et al.2013). It becomes clear that Mov10 inhibits virus infection by enhancing RIG-I-MAVS-Independent IFN Induction (Cuevas et al.2016) and stabilizing A3G from degradation (Chen et al.2017).
The anticipated evidence of viral disruption of PB also came from the study with poliovirus (PV), a plus-strand RNA virus showing that PB are disrupted during PV infection in cells by 4 h post infection (Dougherty et al.2011). This function is attributed to viral proteinase 3C which degrades several components of PB including Xrn1 and Dcp1a, but not affecting others such as GW182, Edc3 and Edc4. Rotaviruses disassemble PB by using viral RNA as a sponge for RNA binding proteins to redistribute several PB components, including Ago2, GW182 and Dcp1 PB (Oceguera et al.2018). In fact, rotavirus disrupts PB through multiple mechanisms. The viral NSP1 protein seems to degrade PB component Pan3, while relocalizing other two components (Xrn1 and Dcp1a) (Bhowmick et al.2015). Intriguingly, exclusion of SG and PB components from the viroplasm is important for rotavirus replication and progeny virus production (Dhillon and Rao 2018).
-
While RNA viruses have evolved to co-opt or modulate the assembly of PB, this effect is rather unclear during infection by DNA viruses. Since most of the DNA viruses replicate and assemble in the nucleus, therefore as proposed for RNA viruses, accumulation of viral RNAs in PB for assembly cannot be a strategy required by DNA viruses. However, the close relationship of PB with translational repression reasonably provides a foundation for PB being antiviral cellular components against DNA viruses. Thus it is assumed that those factories suppressing mRNA translation would inhibit protein production of DNA viruses. To fight back, the DNA viruses have to develope strategies to bypass this antagonism mediated by PB for their survival and productive infection (Table 2).
Adenovirus E4 11 k, the product of E4 ORF3, accumulates viral late mRNA transcripts and at least five proteins of PB (Rck/p54/DDX6, Ago2, xrn1, Ge1, and Lsm-1) in the E4 11 k-induced cytoplasmic aggresomes. Redistribution of the PB components to the aggresomes, not to the PB, leads to inactivate or destroy these proteins. E4 11 k protein interacts with RNA helicase DDX6, one of the PB proteins, for its redistribution. Because PB are the sites for mRNA degradation, their alteration by E4 11 k suggests a role of E4 11 k in viral late mRNA accumulation (Greer et al.2011).
The role of PB in regulation of cytomegalovirus infection remains elusive. First, HCMV infection does not affect, but rather accumulates the formation of PB; second, PB formed during HCMV infection do not contain Ago2; third, HCMV prevents viral IE1 mRNA, a major IE gene product to encode a critical protein for viral gene expression and replication, from colocalization with PB (Seto et al.2014).
By generating a transgenic mice deficient of PB component LSm14A (or Rap55), recent studies showed that LSm14A plays a critical and specific role in the induction of antiviral cytokines (IFN-β, IFN-α, and IL-6) in dendritic cells (DCs). DNA viruses (HSV-1 and murine herpesvirus 68) and RNA virus VSV trigger this induction, but Sendai virus lacks such an effect (Anderson and Kedersha 2009; Liu et al.2016). LSm14A deficiency specifically downregulates MITA/STING (stimulator of interferon genes) level in DCs by impairing its nuclear mRNA precursor processing. In contrast to its role in mRNA decay, this study revealed a role of LSm14 in nuclear mRNA precursor processing and cell-specific regulatory mechanism of antiviral immune responses (Liu et al.2016).
KSHV kaposin B, a latent protein linked with cancer progression, induces PB dispersion (Corcoran et al.2015). Kaposin B activates the stress-responsive kinase MK2 in endothelial cells (ECs) to selectively block the decay of AU-rich mRNAs (ARE-mRNAs) which encode pro-inflammatory cytokines and angiogenic factors and to reprogram ECs through post-transcriptional control of EC gene expression and secretion. KSHV ORF57 protein inhibits the formation of PB during lytic infection by disrupting the essential interaction of Ago2 with GW182 (unpublished data). These data provide the first evidence that a tumor virus RNA-binding protein ORF57 antagonizes the RNA regulatory pathway of host antiviral defenses during lytic infection.
Assembly of P-Bodies (PB)
RNA Viruses and PB
DNA Viruses and PB
-
SG are highly dynamic structures (Jain et al.2016), which constantly exchange their components to regulate gene expression and are thought to be antiviral. SG composition appears to vary according to the inducing stimulus (Table 3). It's clear that SG assembly/disassembly is a tightly regulated process which accompanies rearrangements of RNA and proteins (Wheeler et al.2016). Although significant advances have been made to understand how viruses regulate SG formation, our current knowledge is not suffucient to fully elucidate the machanism how SG are regulated in living cells. Further works are needed to address the following questions: First, is there any pathway to be a target for antiviral drug development? Second, do SG function as platforms that potentiate virus recognition? Third, is any unexplored pathway leading to SG formation which could be visualized by fluorescence in situ hybridization techniques—including single molecule RNA tracking methods in combination with super-resolution microscopy? Using viruses as a research tool will definitely teach us how the host fights virus infections and how the viruses get away from its host resistance.

Table 3. Viruses and SG components
PB affect viral infections in multiple ways. Thus, it is difficult to generalize a common viral strategy in a particular virus group to interact with the components of PB. The noticed evidence is that viruses in the same family may show extremely distant behavior when they come to interact with PB (Table 2). More studies on virus interactions with PB will be required to characterize the PB to be proviral or antiviral in a context-dependent manner. Other key questions in the field for future studies are: (1) to understand the mechanisms that regulate PB formation in cells. Viral manipulation of PB may provide a better platform to understand this regulation; (2) to determine which viral RNA species preferentially travel through these RNA granules and which ones do not? (3) to identify the RNA elements dictating viral RNA to escape from SG and PB. Thus, discovery of virus regulations of PB assembly represents a new paradigm of virus-host interactions.
-
This work was supported by grants from the China Natural Science Foundation (81825015 and 31630086), the Natural Science Foundation of Hubei Province Innovation Group (2017CFA022), and Intramural Research Program of NCI/NIH (1ZIASC010357 to ZMZ).
-
The authors declare that they have no conflict of interest.
-
This article does not contain any studies with human or animal subjects performed by any of the authors.
-
This article is distributed under the terms of the Creative Commons Attribution 4.0 International License (http://creativecommons.org/licenses/by/4.0/), which permits unrestricted use, distribution, and reproduction in any medium, provided you give appropriate credit to the original author(s) and the source, provide a link to the Creative Commons license, and indicate if changes were made.

 DownLoad:
DownLoad: