














HTML
-
Hepatitis C virus (HCV), a major human pathogen, is responsible for the development of chronic liver diseases, including steatosis, fibrosis, cirrhosis and hepatocellular carcinoma (Chisari 2005; Sanyal et al. 2010). All HCV proteins interact with host cell membranes, either directly as membrane-binding proteins or, in the case of NS3, via interaction with the membrane-anchoring protein, NS4A. HCV has two envelope glycoproteins: both proteins are heavily glycosylated on the N-terminal ectodomain in the lumen of the endoplasmic reticulum (ER) (Op De Beeck et al. 2001). Furthermore, they contain transmembrane domains (TMD) within the C-terminal ends (Op De Beeck et al. 2004), which co-localizes with all other nonstructural proteins on ER-derived membranes. During HCV assembly, E1 and E2 interact non-covalently and form heterodimers, which are inserted into enveloped lipid membrane of the viral particle.
HCV NS2 is required for HCV polyprotein processing and particle assembly. It is a 217-amino acids (aa) long cysteine-protease composed of a highly hydrophobic N-terminal membrane binding domain (aa 1-93) and a C-terminal globular and cytosolically oriented protease subdomain (aa 94-217) (Lorenz et al. 2006; Lange et al. 2014). The membrane domain is composed of three predicted transmembrane domains and serves as a molecular platform for the coordination of HCV structural and nonstructural proteins in viral assembly (Jirasko et al. 2010; Ma et al. 2011; Popescu et al. 2011; Stapleford and Lindenbach 2011; Guo et al. 2015). The C-terminal is capable of forming a homodimeric cysteine autoprotease with twin composite active sites composed of two residues from one chain and one residue from the other, that is responsible for cis cleavage at the NS2-NS3 junction within its C-terminal domain (Santolini et al. 1995; Yamaga and Ou 2002; Lorenz et al. 2006), and NS3-4A processes the remainder of the nonstructural proteins (Lindenbach and Rice 2001).
HCV NS5A is an RNA binding phosphoprotein composed of three domains (Domains Ⅰ, Ⅱ and Ⅲ) that are separated by low complexity sequences (LCS Ⅰ and LCS Ⅱ) and an N-terminal amphipathic alpha-helix (Brass et al. 2002; Penin et al. 2004; Tellinghuisen and Rice 2005; Appel et al. 2008). This anchor helix is necessary and sufficient to target NS5A or a heterologous fusion protein to the endoplasmic reticulum (ER) or an ER-derived modified compartment by a post-translational mechanism, resulting in an integral membrane protein (Brass et al. 2002; Penin et al. 2004). Domain Ⅰ and Domain Ⅱ played an essential role in HCV replication but hardly participate in HCV particle production. In contrast, Domain Ⅲ can be deleted or replaced by green fluorescent protein with no dramatic effect on RNA replication (Moradpour et al. 2004; Appel et al. 2005; Han et al. 2009). It has been reported that C-terminal serine cluster of domain Ⅲ has a crucial role in HCV infectious particle assembly (Appel et al. 2008). Two NS5A phospho variants are found, designated a basally phosphorylated form of 56 kDa and a hyperphosphorylated form of 58 kDa according to their apparent molecular weights (Kaneko et al. 1994; Tanji et al. 1995). Each phosphorylation status of NS5A interacts with multiple host and viral protein partners (Huang et al. 2007) and executes a key regulatory role in the switch between replication and infectious virus assembly (Appel et al. 2008; Tellinghuisen et al. 2008; Reiss et al. 2013).
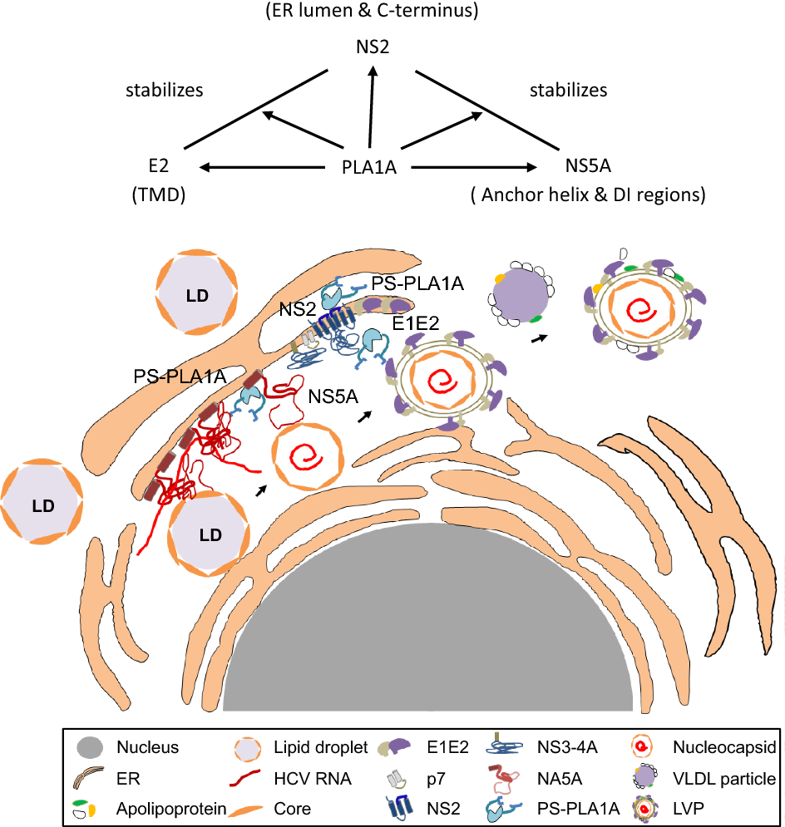
PLA1A is a member of the phospholipase family. Unlike other members of the lipase family, PLA1A has a shorter lid (12 residues) and a shorter beta-9 loop (13 residues). The shorter loops in PLA1A gene may result in the gene acquiring various lipase activities or host functions. It should be noted that PLA1A cleaves fatty acids at the sn-1 position of both phosphatidylserine (PS) and 1-acyl-2-lysophosphatidylserine (lysoPS) (Winkler et al. 1990; Nagai et al. 1999). In a previous study, we showed that the PLA1A could interact with the HCV E2, NS2 and NS5A proteins and facilitate NS2-E2 and NS2-NS5A complex formation during virus assembly (Guo et al. 2015).
In this study, we showed that the structural determinants of PLA1A, E2, NS2 and NS5A proteins are important for HCV assembly by providing a detailed characterization of various PLA1A and HCV protein mutants. These results reveal a complex mechanism of the organization of PLA1A in HCV assembly and shed light on the contribution of PLA1A to complex formation of NS2-E2 and NS2-NS5A in HCV assembly.
-
Human liver tissue samples from fine-needle biopsies were obtained from HCV-infected patients. Normal human liver tissue samples were obtained from either spare donor tissues intended for transplantation or from normal liver tissues resected from patients with benign hepatic tumors. All human tissue samples were collected from the Liver Unit of The First Hospital of Jilin University and Tongji Hospital, Tongji Medical College of Huazhong University of Science and Technology. Diagnosis of patients with chronic HCV infection and analysis of all biopsies were based on standard serological assays and the presence of abnormal serum aminotransferase concentrations for at least 6 months. All patients with HCV tested positive for HCV antibody based on a third-generation ELISA test. HCV infection was confirmed by detection of circulating HCV RNA using an HCV PCR-based assay (Qiagen, Hilden, Germany). At the time of biopsy, liver tissue (2-3 mm) was immediately frozen in TRIzol and stored at -80 ℃.
-
Extraction of intracellular metabolites and nuclear magnetic resonance (NMR) analysis were performed (Li et al. 2015).
-
Human embryonic kidney 293T cells, African green monkey kidney epithelial Vero cells and the derivative cell lines, Huh-7.5.1, from human hepatoma Huh7 cells, were maintained in Dulbecco's Modified Eagle Medium (DMEM) (Invitrogen, Carlsbad, USA) supplemented with 2 mmol/L L-glutamine, nonessential amino acids, and 10% Fetal Bovine Serum (FBS) (Invitrogen), 100 U/mL penicillin and 100 μg/mL streptomycin at 37 ℃ in a 5% CO2 incubator (Xu et al. 2012).
-
A complete list of primers is provided in Supplementary Table S1. The HA- or Flag-tagged expression plasmids of HCV E2, NS2 and NS5A of genotype 2a (JFH1, AB047639) and the pXJ40-HA-PLA1A was previously described. All deletion mutations and site-directed mutagenesis of PLA1A, E2, NS2 or NS5A were amplified and inserted into the pXJ40-HA or pXJ40-Flag vectors. A different pair of split mCherry fusion constructs for visualizing protein-protein interactions were amplified and inserted into the pMC160 or pMN159 vectors (Fan et al. 2008). pMC160 vector was digested by Hind Ⅲ and Kpn Ⅰ pMN159 vector was digested by Hind Ⅲ and Pst Ⅰ. The wild type or deletion mutation coding sequence of PLA1A, E2, NS2 and NS5A was digested by the same restriction enzymes and inserted into the corresponding sites of pMC160 or pMN159.
-
The lentiviral vector pWPI-Puro has been used for cloning of pWPI-hPLA1A plasmid for the generation of stable cell lines under puromycin selection. The PLA1A (NM_001206960) coding sequence was amplified by PCR and inserted into the pWPI-Puro plasmids. The lentiviruses were produced in 293T cells by co-transfection of the pWPI-Puro or pWPI-hPLA1A, pCMV-dR8.91 and pMD2.G (Addgene, http://www.addgene.org). The lentivirus-containing supernatants were harvested at 72 hpt. To generate the PLA1A overexpression cell line, the Huh- 7.5.1 cells were transduced with the lentiviruses in the presence of 20 μg/mL polybrene (Sigma), and stable overexpressing pools were isolated by puromycin selection and named Huh-7.5.1-hPLA1A.
-
293T cells were transfected with the different pXJ40-based constructs by using Lipofectamine 2000 (Invitrogen). Cells were seeded into 60 mm culture dish, 1 day prior to transfection. For transfection, 10 μg plasmid was mixed with 15 μL Lipofectamine 2000 and applied to cells as recommended by the manufacturer. After 2 days, cells were harvested for coimmunoprecipitation (co-IP) assay.
-
The HCV J399EM strain was derived from the JFH-1 virus by insertion of eGFP into the HCV NS5A region (Han et al. 2009). For HCV infection, Huh-7.5.1 cells were incubated with 0.1 MOI HCV J399EM virus for 6 h at 37 ℃. Then cells were rinsed three times with phosphatebuffered saline (PBS) and maintained in new medium to indicate time points. The absolute titer values of infectious HCV particles in culture supernatants were determined by limiting dilution analysis (Randall et al. 2006).
-
Total RNA from cultured cells and HCV RNA in the supernatant were extracted using TRIzol reagent (Invitrogen) and TRIzol LS reagent (Invitrogen) according to the manufacturer's protocols, respectively. Specific mRNAs and HCV RNAs were quantified by one-step real-time RTPCR using the QuantiFast SYBR Green RT-PCR kit (Qiagen). The levels of mRNAs or HCV RNAs were normalized to the levels of beta-actin with the standard curve method. The forward and reverse primers used to amplify PLA1A was previously described (Guo et al. 2015); the primers for HCV and actin were previously described as well (Zhu et al. 2014).
-
The following antibodies were used for Western blot, immunoprecipitation and immunofluorescence. Mouse monoclonal antibodies against Flag (#F1084, Sigma), mouse monoclonal antibodies against HA (#H9658, Sigma), rabbit polyclonal antibodies against Flag (#2368S, Cell Signaling Technology), rabbit monoclonal antibodies against HA (#3724, Cell Signaling Technology), rabbit polyclonal antibodies against PLA1A (Abmart), mouse monoclonal antibodies against anti-ApoE (#Ab1906, Abcam) and mouse monoclonal antibodies against betaactin (#sc-47778, Santa Cruz). The proteins were visualized using suitable HRP-conjugated secondary antibodies (Jackson Immuno Research). The Alexa Flour 561/488/ 633-conjugated secondary antibodies used for indirect immunofluorescence staining were obtained from Invitrogen.
-
Cells for both Western blotting and immunoprecipitation were lysed in immunoprecipitation (IP) buffer containing 50 mmol/L Tris (pH 7.5), 1 mmol/L EGTA, 1 mmol/L EDTA, 1% Triton X-100, 150 mmol/L NaCl, 100 μmol/L phenylmethylsulfonyl fluoride (PMSF), and a protease inhibitor cocktail (Complete Mini, Roche) for 30 min (Xu et al. 2012). Cell lysates were centrifuged at 14, 000 ×g for 1 min at 4 ℃ and quantified using the Bradford method (#500-0006, BioRad). For Western blotting, the supernatants were recovered and boiled in loading buffer. For immunoprecipitation, the supernatants were recovered and mixed with 2 μg of primary antibody per 1 mg protein samples and then incubated overnight at 4 ℃. The reaction mixtures were then mixed with protein G agarose (#2549373, Millipore) and incubated for an additional 2 h at 4 ℃. Protein G agarose-bound immune complexes were collected by centrifugation at 14, 000 ×g for 1 min, washed at least five times with IP buffer, and boiled in loading buffer. Then the samples were centrifuged at 14, 000 ×g for 1 min and loaded onto 12% SDS-polyacrylamide gels. The proteins were separated at a constant voltage (120 V), then the gels containing proteins were transferred onto a nitrocellulose filter membrane (Millipore). Membranes were blocked by incubation with 5% nonfat milk, and proteins were detected by using primary antibodies and secondary antibodies conjugated with horseradish peroxidase. The proteins were visualized using suitable HRPconjugated secondary antibodies (Jackson Immuno Research) and SuperSignal-Femto chemiluminescent substrate (Pierce).
-
The Vero cells were plated in 35-mm tissue culture dishes at 70%-80% confluence, 1 day before transfection. The cells were transfected with a pair of split mCherry fusion constructs, using Lipofectamine 2000 reagent (Invitrogen; USA) according to the manufacturer's instructions. Transfected cells were incubated at 37 ℃ (5% CO2) for 12 h and continued at 25 ℃ overnight (5% CO2) before imaging. Twenty-four hours after transfection, cells were fixed and stained with DAPI. The signal representing interaction was analyzed by confocal microscope.
-
Huh-7.5.1 cells were infected with infectious HCV J399EM virus at MOI 0.1 for 48 h. Subsequently, normal rabbit IgG (4 μg), ApoE- and PLA1A-specific antibody were each mixed with 1 mL HCVcc (cell culture produced HCV) supernatant and rotated overnight at 4 ℃. After incubation, 60 μL washed protein G-conjugated agarose beads (#2549373, Millipore) was added to each group, and sequentially, the samples were rotated for 4 h at 4 ℃. Unbound HCV particles were removed by at least five washes in IP buffer as described above. HCV particlesantibody-beads complex were resuspended into 250 μL nuclease-free water, and HCV RNA was extracted with 750 μL TRIzol LS reagent (#10296-028, Invitrogen). HCV RNA copy numbers were quantified by one step real-time RT-PCR as described above. The groups of rabbit IgG and ApoE-specific antibody served as negative and positive controls, respectively.
-
Immunofluorescence protocol was performed as described elsewhere (Xu et al. 2012). In brief, Huh7.5.1 cells were grown on glass coverslips and electroporated with transcribed HCV J399EM RNA in vitro. At various time points after electroporation, Huh7.5.1 cells were fixed, permeabilized and blocked. Samples were incubated overnight with the primary antibodies in various combinations. Alexa Fluor 561/488/633-conjugated secondary antibodies (diluted 1:1000; Invitrogen) were added for 1 h. Then the coverslips were washed three times with PBS and stained with DAPI (Invitrogen). Images of the samples were taken using an UltraView Vox confocal microscope (Perkin Elmer, USA).
-
Differences between groups were evaluated using the twotailed, unpaired Student's t test (GraphPad Software, Inc., La Jolla, CA). Coprecipitation efficiency and fluorescence images were analyzed using Image J (National Institutes of Health, USA). P-values were calculated, and statistical significance was reported as highly significant with *(P < 0.05). Analytic results are presented as mean ± SD.
Patients and Biopsies
Metabolite Profiling
Cell Culture
Primers and Plasmid Constructs
Stably Overexpressing PLA1A Cell Line Construction
Transient Transfection of DNA Expression Constructs
Virus and HCV Infection
Quantitative Real-time RT-PCR
Antibodies
Western Blotting and Immunoprecipitation
The mCherry-based BiFC Assay
Immunoprecipitation of HCV Particles
Immunofluorescence Analysis and Confocal Microscopy
Statistical Analysis and Software
-
In previous studies, we had identified that PLA1A expression was enhanced by 20 fold in JFH-1 infected cells (Guo et al. 2015). To confirm the results, we examined PLA1A expression in liver biopsy samples from HCV-infected patients. The demographic and clinicopathological characteristics of 42 biopsies obtained from HCV-infected patients and 10 biopsies from normal control patients included in the study are shown in Supplementary Table S2. We observed a statistically significant increase in PLA1A expression in liver biopsies from HCV-infected patients (Fig. 1A). In the group of 42 HCV-infected patients, PLA1A mRNA expression levels were positively associated with HCV viral loads in the liver or the serum (Fig. 1B). The multivariate logistic regression analysis showed that PLA1A mRNA level and HCV RNA in serum (OR = 9.037; CI 95% 1-36, P < 0.01) or HCV RNA in liver (OR = 43.18; CI 95% 1-36, P < 0.001) were dependent variables associated with HCV infection. To gain insight into the potential metabolic signature associated with PLA1A overload, we examined the influence of HCV infection on the metabolites involved in PS from HCV-infected cells using ELISA. The metabolic profile of HCV-infected cells demonstrated a clear shift toward lysoPS compared with mock-infected counterparts in 72 hpi. HCV-infected cells, which showed lower levels of PS and produced more cellular or secreted Lyso-PS (18:0), which are the substrate or product of PLA1A (Fig. 1C). These results were in agreement with a previous report on the increased expression of PLA1A in HCV life cycle and demonstrated that PLA1A was upregulated in vivo.
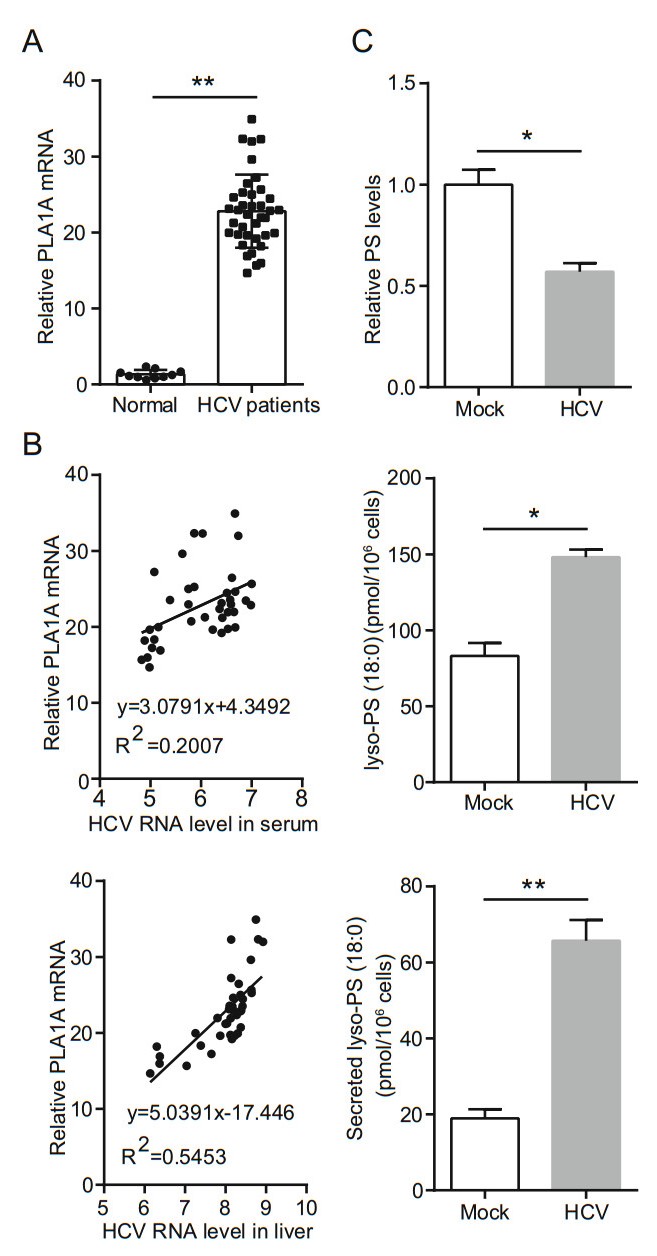
Figure 1. HCV infection upregulates host PLA1A expression. A Box plot diagrams show the PLA1A mRNA levels in liver biopsies of 10 normal control patients and 38 patients with chronic hepatitis C. B There is a positive correlation between PLA1A mRNA and HCV virus load in serum or liver. C Levels of phosphatidylserine (PS), lyso-PS (18:0), and secreted lyso-PS (18:0) at the indicated time points. Data are presented as means ± SDs. *P < 0.05; **P < 0.001.
-
A previous report has highlighted the importance of the interaction of PLA1A with HCV E2, NS2 and NS5A proteins for the assembly and release of infectious HCV particles (Guo et al. 2015). This result was perplexing due to the membrane topologies of the proteins. So we aimed to investigate structural determinants of PLA1A, E2, NS2 and NS5A proteins important for HCV assembly by providing a detailed characterization of different PLA1A and HCV protein mutants. Firstly, using HCV FLAG-tagged E2 (FLAG-E2), FLAG-tagged NS2 (FLAG-NS2) or FLAGtagged NS5A (FLAG-NS5A) proteins as bait, interactions with endogenously expressed PLA1A were detected by immunoprecipitation and Western blot assay in Huh-7.5.1 cells (Fig. 2A). We further identified the PLA1A-E2, PLA1A-NS2 and PLA1A-NS5A interactions using the mCherry-based red bimolecular fluorescence complementation (BiFC) system (Fan et al. 2008), which is based on fusion proteins with complementary fragments (MC160 and MN159) of the monomeric fluorescent construct. When the MC160 fragments are in close proximity due to the protein-protein interaction, the MN159 fragments form a beta-barrel structure and emit red fluorescence. MC160-mp53-MN159-mLTag and MC160-p53-MN159-Ltag served as negative and positive controls, respectively. Vero cells were transfected by indicated pair of mCherry fusion constructs as shown in Fig. 2B. Thus, the result further represented structures with PLA1A with HCV E2, NS2 and NS5A proteins in close proximity in cells. Furthermore, the result showed structures with PLA1A and E2 in close proximity than NS2 and NS5A, and strongly suggest PLA1A-E2 physical interaction in cells.
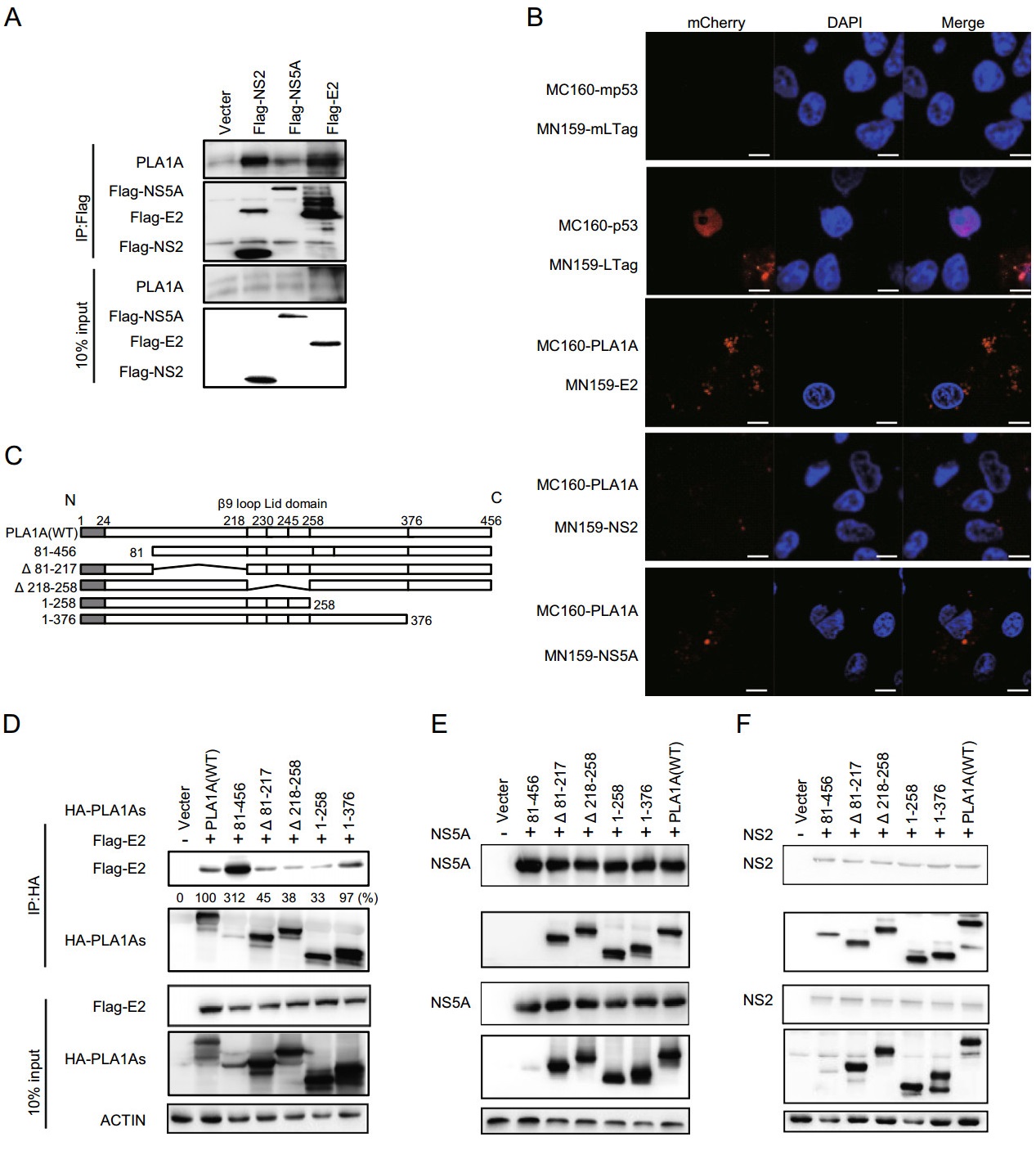
Figure 2. Interaction of PLA1A with E2, NS2 or NS5A. A Huh-7.5.1 cells were transfected with HCV E2, NS2 or NS5A for 48 h, and lysed in immunoprecipitation buffer. Co-immunoprecipitation experiments with anti-PLA1A recognizing endogenous PLA1A, anti-FLAG recognizing E2, NS2 or NS5Awere performed as indicated. B Detection of the PLA1A interaction with E2, NS2 or NS5A in transfected cells using the mCherrybased red BiFC system. Vero cells were transfected by indicated pair of mCherry fusion constructs. Twenty-four hours after transfection, cells were fixed and stained with DAPI, and observed under a confocal microscope. MC160-mp53-MN159-mLTag and MC160-p53-MN159-Ltag served as negative and positive controls, respectively. Scale bars represent 10 μm. C Schematic representation of HA-tagged, deleted in PLA1A proteins used for E2, NS2 and NS5A interaction mapping and subsequent analyzes. Residues encompassing the respective deletions are indicated at the left. D-F 293T cells were transfected with plasmids encoding the different deletionswithinHA-taggedPLA1Awith orwithout Flag-tagged E2, NS2 or NS5A of genotype 2a (JFH-1), as indicated at the top. An empty plasmid was used as a negative control. Cell lysates subjected to immunoprecipitation using anti-Flag were subsequently analyzed by Western blot with indicated antibodies. 10% cell lysates were used for normalization of input levels. Numbers at the bottom indicate the coprecipitation efficiency of E2, NS2 or NS5A with individual deletions compared to PLA1A wt and normalized to HCV proteins input levels. Data presented here is representative of three independent experiments.
To determine the specific sequences of PLA1A interaction with E2, NS2 or NS5A in mammalian cells, we constructed various deletions within HA-tagged PLA1A (HA-PLA1As; Fig. 2C). FLAG-E2, FLAG-NS2 or FLAGNS5A was co-expressed in 293T cells together with HAPLA1As, respectively. Subsequently, cell lysates were submitted to co-immunoprecipitation and Western blot with indicated antibodies. The result showed that all mutants of PLA1A still could interact with E2 (Fig. 2D), NS2 (Fig. 2E) and NS5A (Fig. 2F), suggesting multiple regions of PLA1A involved in their interaction.
-
Since E2 interacts with PLA1A, we tried to determine the region in E2 that is responsible for PLA1A binding. We constructed various truncation mutants of E2 (Fig. 3A) and the binding domain was determined by co-immunoprecipitation and Western blot assay. Figure 3B shows that PLA1A binds to E2 mutant harboring carboxyterminal end but not with mutant harboring endoplasmic reticulum lumen region, suggesting that the E2 region spanning aa 336-367 is involved in its interaction with PLA1A. The amount of PLA1A coprecipitating with E2 was quantified and normalized to PLA1A input levels. It is remarkable that the C-terminal transmembrane domain (TMD) of E2 that serves as a membrane anchor was required for the interaction with PLA1A, as E2 mutant aa 1-272, lacking this TMD, similarly loses its ability to bind PLA1A (Fig. 3B). We further identified the above interaction by the mCherry-based red BiFC system. Co-expression of MC160-E2 or MC160-E2/1-336 and MN159-PLA1A fusion proteins in Vero cells, showed only MC160-E2 and MN159-PLA1A reconstituted red cellular fluorescence as shown in Fig. 3C. The percentage of cells with the brilliant redness was counted and shown in Fig. 3D. The finding suggested that the TMD region of E2 is involved in the interaction with PLA1A.
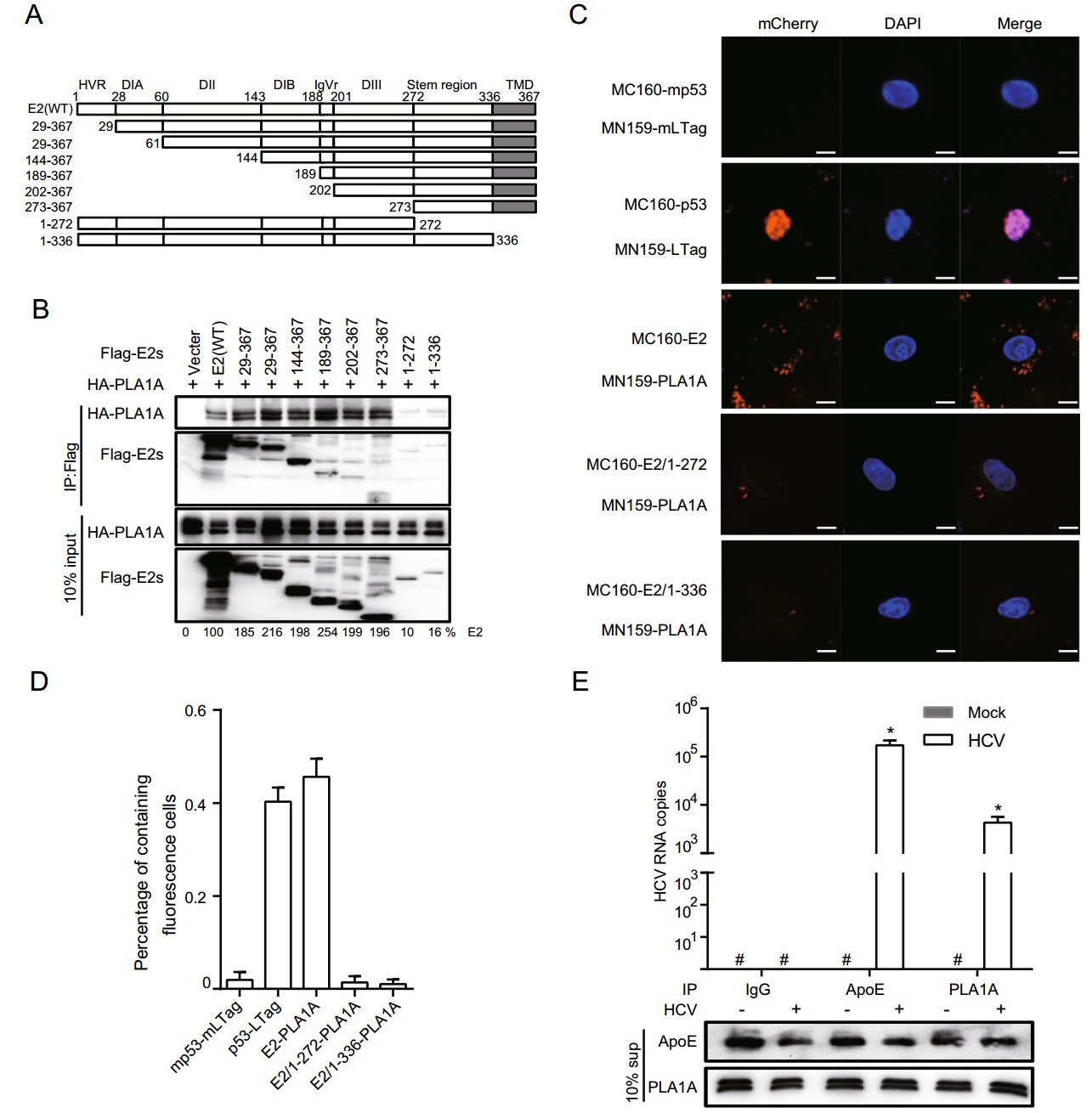
Figure 3. Characterization of E2 binding to PLA1A. A A schematic representation of the HCV E2 domain and deletion mutations. B 293T cells were co-transfected with the Flag-tagged E2 expression plasmid in the presence of HA-PLA1A expression plasmid, as indicated at the top. An empty plasmid was used as a negative control. Cell lysates of the transfected cells were immunoprecipitated with anti-Flag antibody. The resulting precipitates and whole cell lysates used in immunoprecipitation (IP) were examined by immunoblotting using anti-FLAG- or anti-HA antibody. 10% cell lysates were used for normalization of input levels. C Detection of the PLA1A-E2 interaction in Vero cells using the mCherry-based red BiFC system. Scale bars represent 10 μm. D The percentage of cells with brilliant redness. E Huh-7.5.1 cells were infected with infectious HCV J399EM virus at MOI 0.1 for 48 h. Subsequently, the cells supernatants (sup) from the control cells or HCV infection cells were subjected to coprecipitate using anti-ApoE antibody (1:100 dilution), anti-IgG (1:100 dilution) or anti-PLA1A antibody (1:100 dilution). Total RNA and protein were extracted from the coprecipitated samples and analyzed for HCV RNA by quantitative RT-PCR. The Western blot indicated the standardization of the sample. Rabbit-IgG and ApoE antibody served as negative and positive controls, respectively. Results presented are representative of three independent experiments.
Although PLA1A is a secreted protein and the lipase activity has been verified, its extracellular functions still remain unknown in HCV infection. The results strongly suggest PLA1A-E2 physical interaction in cells. Thus, we infected Huh-7.5.1 cells with 0.1 MOI of J399EM for 48 h. Subsequently, the cells supernatant was subjected to coprecipitate using anti-ApoE antibody, anti-IgG or antiPLA1A antibody. Total RNA was then extracted and analysed for HCV RNA by quantitative RT-PCR and the sample were homogeneous by Western blot for relative antibody (Fig. 3E). ApoE associate with mature HCV virions and play an important role in virion infectivity (Jiang et al. 2012; Hueging et al. 2014; Zhu et al. 2014). Rabbit-IgG antibody served as negative control. This result suggested that PLA1A may participate in the formation of HCV mature particles.
-
To identify the region within NS2 involved in the interaction with PLA1A, we tested different deletion mutants of NS2 for their capacity to bind PLA1A. Full-length FLAGNS2 and its deletion mutants were constructed as recombinant fusion proteins (Fig. 4A). The co-IP and immunoblot assay in 293T cells showed that at least two regions encompassing aa 49-73 between NS2 TMDⅡ and TMDⅢ and C-terminal aa 199-217 of NS2 support interaction with PLA1A (Fig. 4B-4D). The amount of PLA1A coprecipitated with NS2 was quantified and normalized to PLA1A input levels. The data obtained in the IP assay were further confirmed by the mCherry-based red BiFC system. Coexpression of MC160-NS2 or dual deletions of NS2 (∆49-73 and ∆197-217) and MN159-PLA1A fusion proteins in Vero cells, showing that only full length NS2 could reconstitute red cellular fluorescence (Fig. 4E). The percentage of cells with the brilliant redness are shown in Fig. 4F. This result is consistent with the above observation that the C-terminal domain is required for E2 in the interaction with PLA1A. Thus, PLA1A can facilitate the formation of the NS2-E2 complexes (Guo et al. 2015), which was also verified here. These results suggest that the ER lumen and C-terminus regions of NS2 are essential to the interaction with PLA1A.
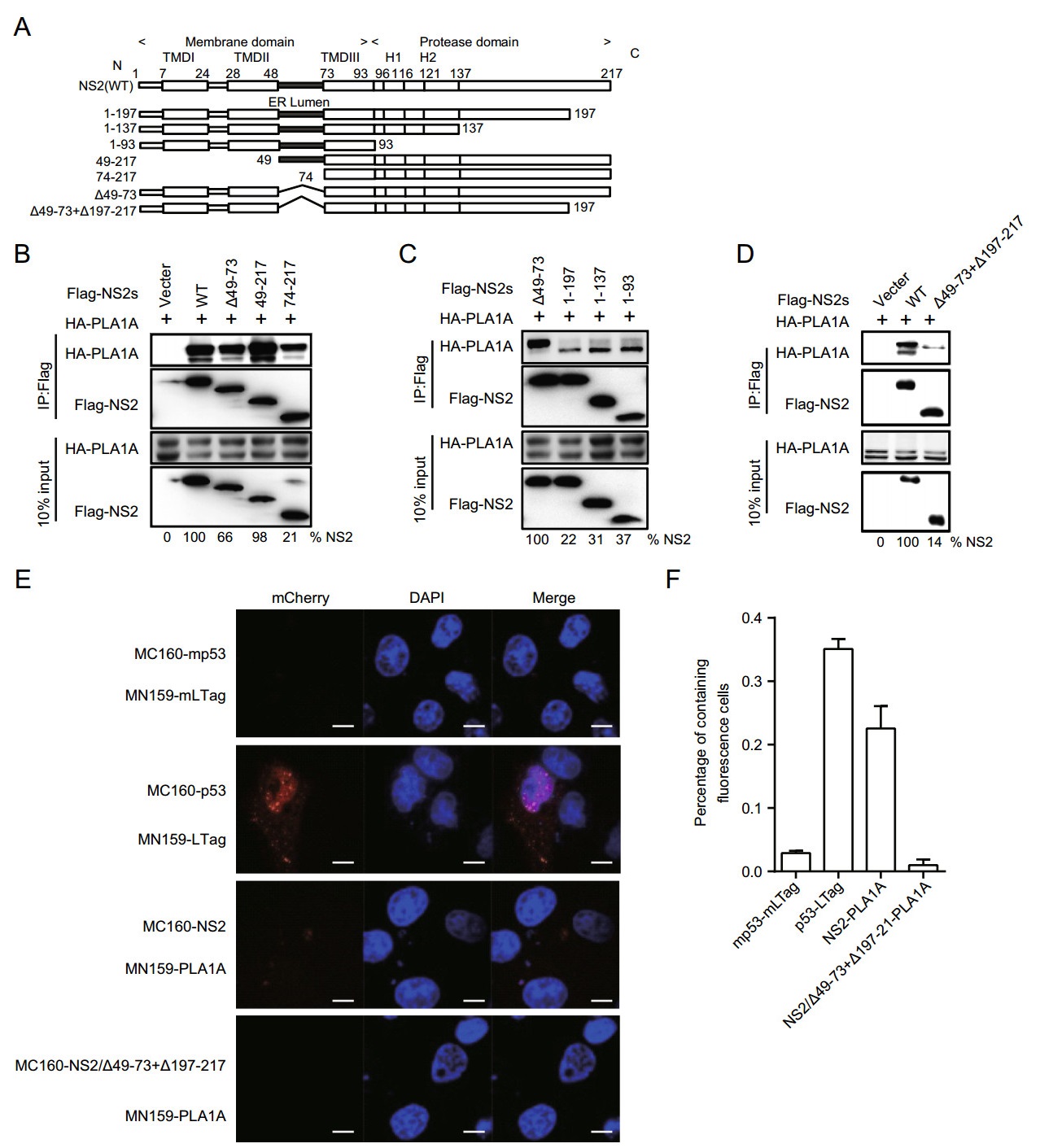
Figure 4. Characterization of NS2 binding to PLA1A. A Schematic representation of each HCV NS2 deletion mutant. B-D 293T cells were transfected with plasmids encoding the different Flag-tagged deletions with HA-tagged PLA1A, as indicated at the top. An empty plasmid was used as a negative control. Numbers at the bottom indicate the coprecipitation efficiency of PLA1A with individual NS2 deletions compared to NS2 wt. E Detection of the PLA1A-NS2 interaction in Vero cells using the mCherry-based red BiFC system. Scale bars represent 10 μm. F The percentage of cells with brilliant redness. Data presented here is representative of three independent experiments.
-
Previously, we found that PLA1A directly interacts with HCV NS5A. To map the NS5A binding region, we designed several deletion mutants within full length NS5A (Fig. 5A). The co-IP and immunoblot assay in 293T cells showed all of mutants with AH depletion could impair PLA1A binding (Fig. 5B).
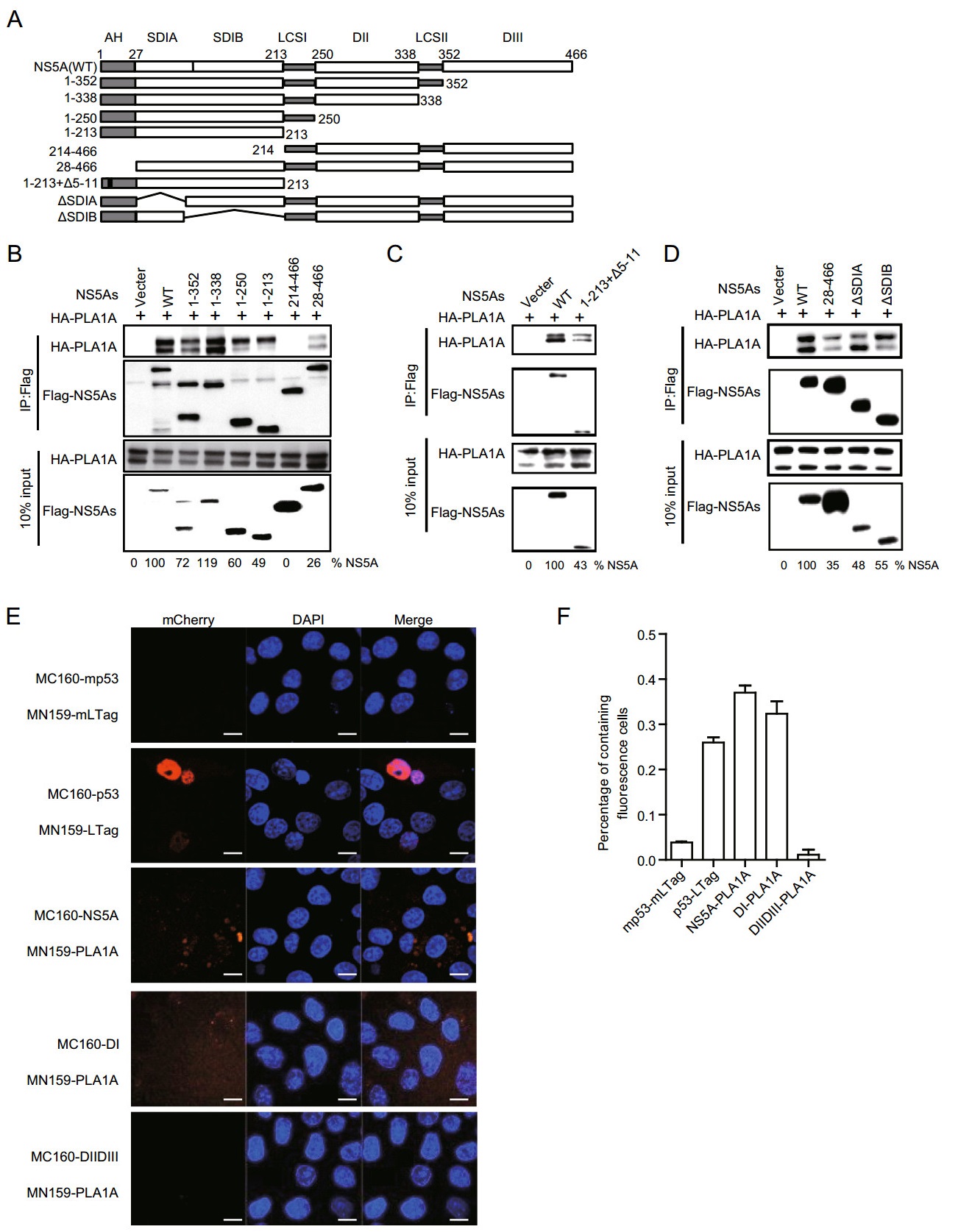
Figure 5. Characterization of NS5A binding to PLA1A. A A schematic representation of domain structure of NS5A and deletion mutations. B-D 293T cells were co-transfected with the different Flag-tagged deletions, NS5A expression plasmid in the presence of a HA-PLA1A expression plasmid, as indicated at the top. An empty plasmid was used as a negative control. Cell lysates of the transfected cells were immunoprecipitated with anti-Flag antibody. Numbers at the bottom indicate the coprecipitation efficiency of PLA1A with individual NS5A deletions compared to NS5A wt. E Detection of the PLA1ANS5A interaction in Vero cells using the mCherry-based red BiFC system. Scale bars represent 10 μm. F The percentage of cells with brilliant redness. Data presented here is representative of three independent experiments.
To determine that the AH region binding to PLA1A was sequences or structures dependent, we constructed a NS5A mutant (1-213 + ∆5-11) which deleted two α-helix turns within the membrane anchor domain and examined its binding ability with PLA1A. Figure 5C shows that NS5A (aa 1-213 + ∆5-11) receded the interaction with PLA1A, which indicated that the AH region played important roles in the interaction of NS5A with PLA1A. PLA1A is a glycosylated protein, the amino acid sequence contains three possible sites for N-linked glycosylation (Junken Aoki et al. 2002). Two PLA1A phenotypes were found and designated a nonglycosylated form of 45 kDa and a glycosylated form of 50 kDa according to their apparent molecular weights. To narrow down the NS5A binding region within DI domain, we constructed two deletion mutants' subdomain Ⅰ A (∆SDI A) and subdomain Ⅰ B (∆SDI B) within full length NS5A. The result showed that∆SDI A and ∆SDI B could bind with two PLA1A forms, respectively (Fig. 5D).
The conclusions were further identified by the mCherry-based red BiFC system. Co-expression of MC160-NS5A, -NS5A DI or -DIIDⅢ and MN159-PLA1A fusion proteins, showing that NS5A and NS5A DI but not with mutant harboring DIIDⅢ reconstituted red cellular fluorescence (Fig. 5E). The percentage of cells with redness was counted and shown in Fig. 5F. Overall, these results suggested that the very N-terminal 28aa and D1 of NS5A contained sequence elements involved in the binding of PLA1A.
-
Our previous reports have indicated that the interaction of NS2 with E2 is independent of PLA1A, but the interaction of NS2 with NS5A could be enhanced by PLA1A. Meanwhile, the NS2-NS5A interaction was important for the assembly of infectious HCV particles (Guo et al. 2015). To further verify whether PLA1A utilized the TMD region of E2 and the AH and DI regions of NS5A to participate in the formation of NS2-E2 and NS2-NS5A complex, we transfected the plasmids containing Flag-tagged wild-type or truncated of E2 or NS5A together with HA-tagged NS2 in the presence or absence of HA-PLA1A in 293T cells. NS2 and PLA1A could be co-immunoprecipitated by FLAG-specific antibody (E2 or NS5A) (Fig. 6A, 6C). The protein-protein interactions were confirmed with reverse immunoprecipitation using HA-specific antibody (Fig. 6B, 6D). Only the NS2 and PLA1A were co-immunoprecipitated by the wild-type E2 or NS5A, suggesting these sequences detected played essential roles in formation of E2-NS2-PLA1A or NS2-PLA1A-NS5A complexes. The above result was further analyzed by BiFC system. Specific enhanced signals indicated the important role of PLA1A (Fig. 6E).
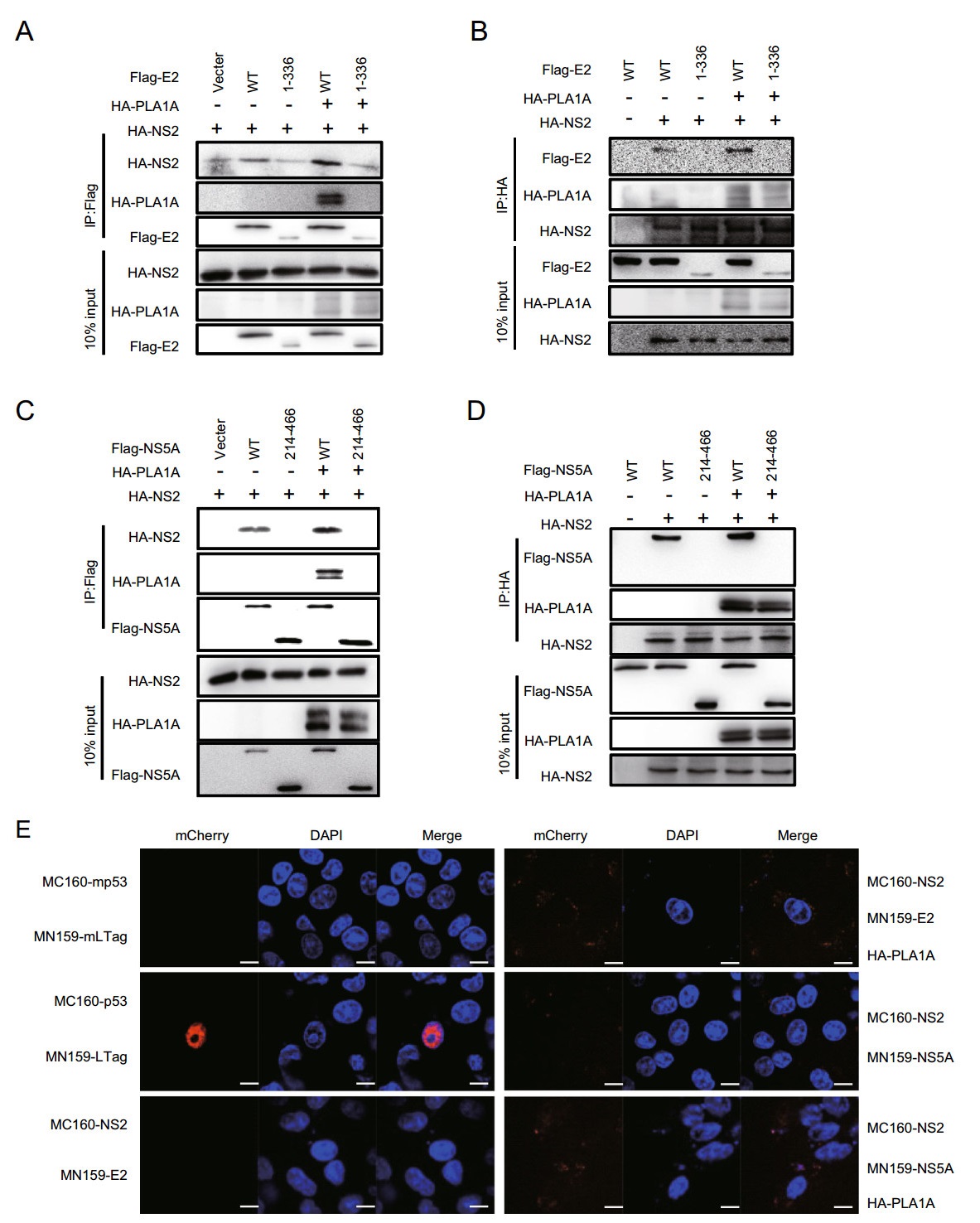
Figure 6. PLA1A coordinates the interaction of NS2-E2 or NS2-NS5A. A-D 293T cells were co- transfected with the Flag-tagged E2 wt, E2-1-336, NS5A wt or NS5A 214-466 expression plasmid and HA-NS2 expression plasmid in the presence or absence of HA-PLA1A expression plasmid, as indicated at the top. An empty plasmid was used as a negative control. Cell lysates of the transfected cells were immunoprecipitated with anti-Flag antibody. The resulting precipitates and whole cell lysates used in immunoprecipitation (IP) were examined by immunoblotting using anti-FLAG- or anti-HA antibody. 10% cell lysates were used for normalization of input levels. Numbers at the bottom indicate the coprecipitation efficiency of NS2 with individual NS5A deletions compared to NS5A wt. E Detection of PLA1A coordinates the interaction of NS2-E2 or NS2-NS5A in Vero cells using the mCherry-based red BiFC system. Scale bars represent 10 μm. Data presented here is representative of three independent experiments.
To determine the specific interaction of NS2 with NS5A, we constructed the deletion mutants of NS2 and NS5A (Fig. 7A, Fig. 7D) and the interaction regions of NS2-NS5A were confirmed with co-immunoprecipitation. Subsequently, cell lysates were immunoprecipitated with antiFlag antibody, suggesting that the NS2 ER lumen region and NS5A AH region were involved in the interaction between NS2 and NS5A (Fig. 7B, 7C, 7E, 7F). These results, together with the above findings, suggest that PLA1A is required for or facilitates the formation of the membrane-associated E2-NS2-PLA1A and NS2-PLA1ANS5A complexes, and NS2-NS5A interact using ER lumen region and AH region.
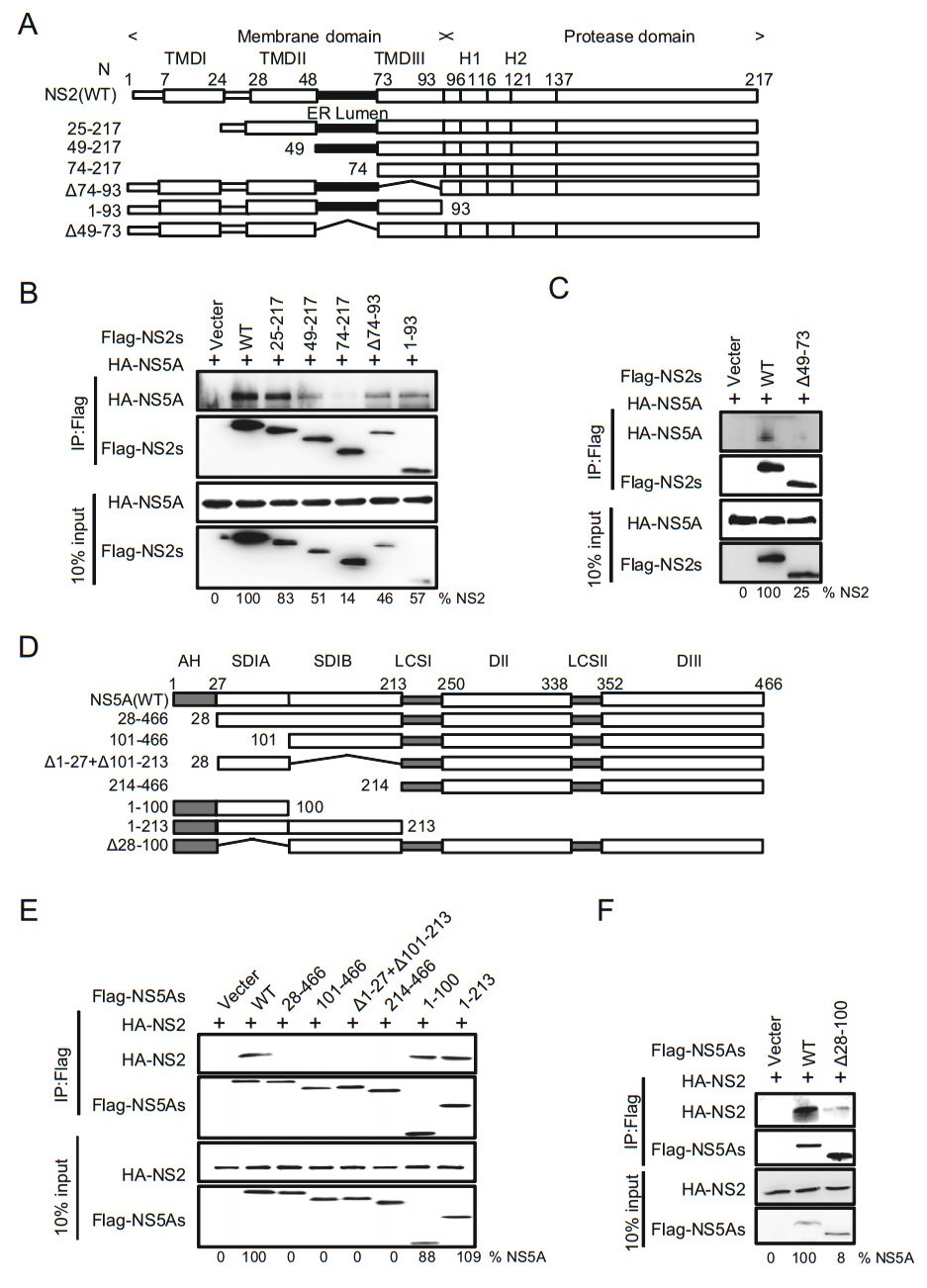
Figure 7. Characterization of NS2 binding to NS5A. A A schematic representation of domain structure of NS2 and deletion mutations. B, C Characterization of NS5A binding to NS2. The indicated Flag-trigged deletion mutants of NS2 and HA-trigged NS5A were co-transfected in 293T cells. An empty plasmid was used as a negative control. Cell lysates of the transfected cells were immunoprecipitated with anti-Flag antibody. The resulting precipitates and whole cell lysates used in immunoprecipitation (IP) were examined by immunoblotting using anti-FLAG- or anti-HA antibody. 10% cell lysates were used for normalization of input levels. D A schematic representation of domain structure of NS5A and deletion mutations. E-F The indicated Flag-trigged deletion mutants of NS5a and HA-trigged NS2 were co-transfected in 293T cells. The experiments were performed as B. Data presented here is representative of three independent experiments.
-
To support and extend the interaction patterns described above with a visual way, we performed colocalization studies of PLA1A with E2, NS2 and NS5A in the HCVcc system. Due to limited endogenous specific antibody of PLA1A, we generated a highly permissive Huh-7.5.1-derived cell pool with stable overexpression of PLA1A (Huh-7.5.1-hPLA1A) for better observation of colocalization of these four proteins with each other (Supplementary Figure S1).
Next the HCV RNA was imported into the control cells or Huh-7.5.1-hPLA1A cells by electroporation and the cells were fixed with antibodies against the PLA1A and HA-tag for indirect immunofluorescence analysis (Fig. 8A). The degree of localization of PLA1A with the viral proteins was quantified (Fig. 8B-8E), each dot in the graph corresponds to one cell. We found that the localization of PLA1A with NS2 was time-independent, but the dynamic localization of PLA1A with NS5A was time-dependent. As above described, the localization of PLA1A with E2 was carried out by the same method. The result suggested that localization of PLA1A with E2 is also timeindependent (Fig. 9A-9D).
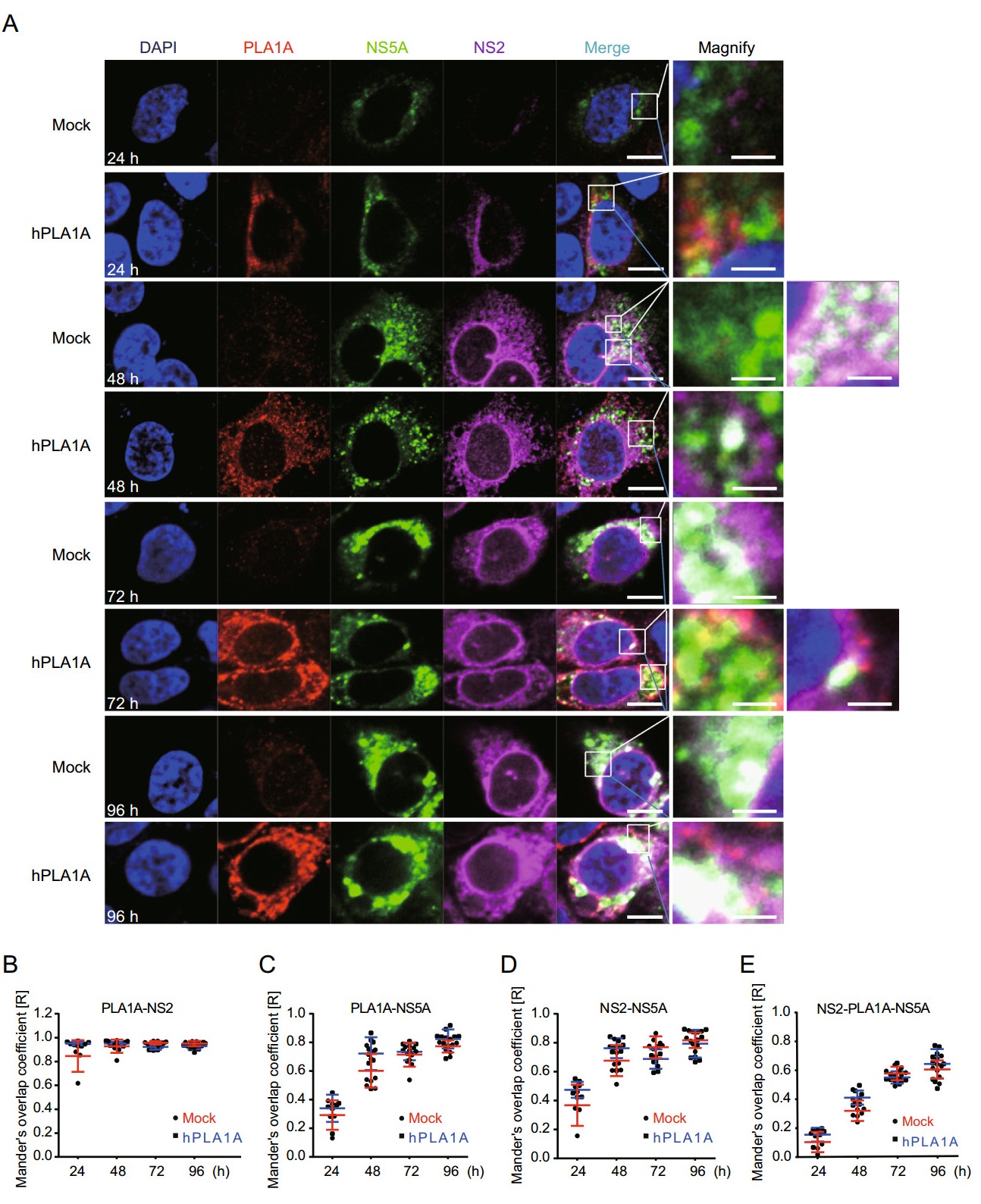
Figure 8. Kinetics of subcellular distribution and colocalization of PLA1A with NS5A and NS2. A Control cells and Huh7.5.1 cells stably overexpressing PLA1A were electroporated with HCV J399EM RNA and at the indicated times post-electroporation cells were fixed and probed with antibodies against the PLA1A and HA-tag for indirect immunofluorescence analysis. NS5A carried green fluorescence protein. The nuclei were stained with DAPI. Scale bars represent 10 μm or 5 μm in regular and magnified images, respectively. B-E The degree of colocalization of PLA1A with given viral proteins was quantified. Each dot in the graph corresponds to one cell.
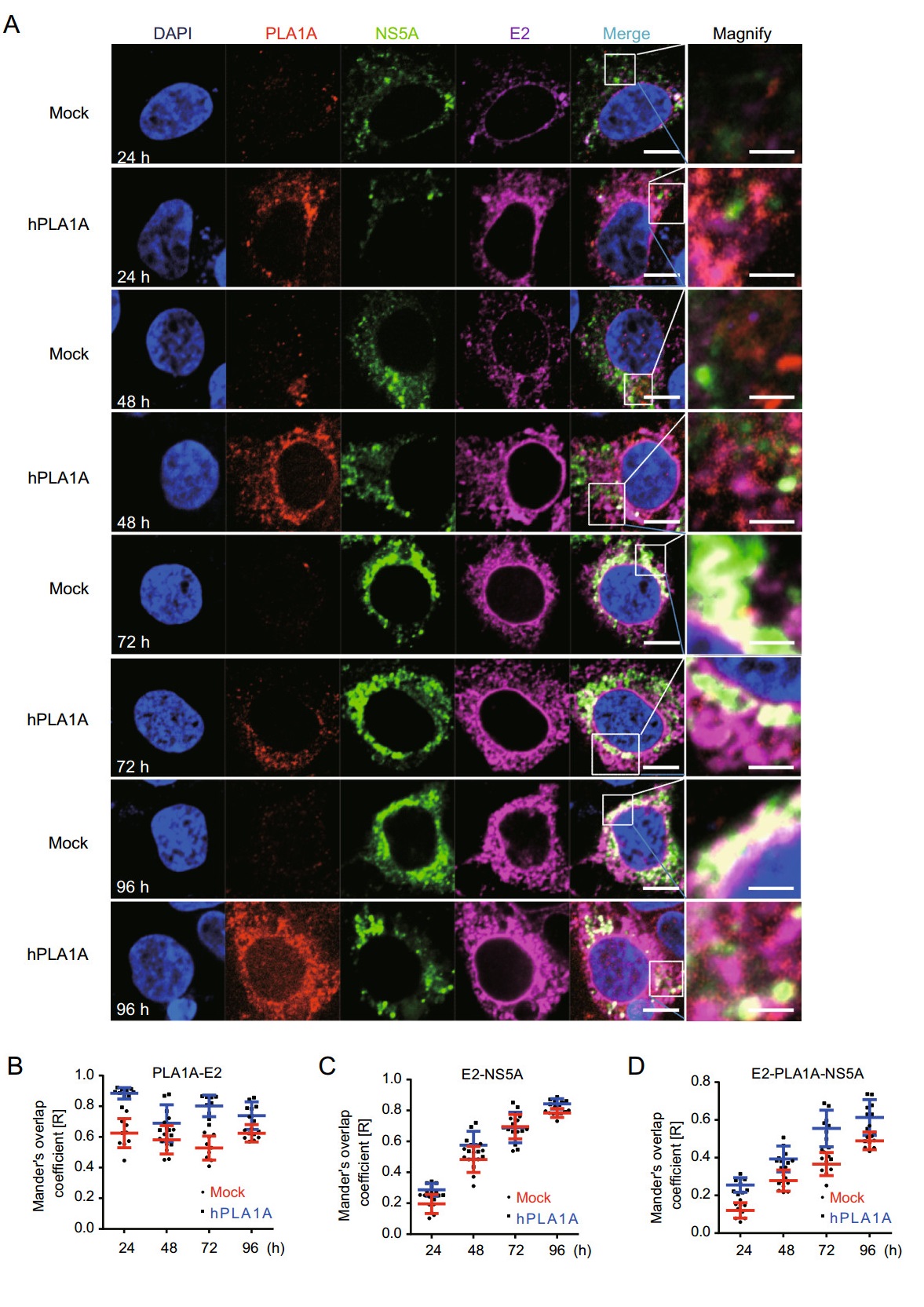
Figure 9. Kinetics of subcellular distribution and colocalization of PLA1A with E2 and NS5A. A Control cells and Huh7.5.1 cells stably overexpressing PLA1A were electroporated with HCV J399EM RNA and at the indicated times post-electroporation, cells were fixed and probed with antibodies against the PLA1A, and E2 for indirect immunofluorescence analysis. NS5A carried green fluorescence protein. The nuclei were stained with DAPI. Scale bars represent 10 μm or 5 μm in regular and magnified images, respectively. B-D The degree of colocalization of PLA1A with given viral proteins was quantified. Each dot in the graph corresponds to one cell.
Taking these findings together, we concluded that the localization of PLA1A with NS2 or E2 is time-independent, but with NS5A was time-dependent change and gradual increase of a strong punctate phenotype was observed among these proteins.
Upregulated PLA1A Expression in HCV-infected Patients
PLA1A Interacts with E2, NS2 and NS5A via Multiple Interaction Sites
E2 Interacts with PLA1A through its Carboxy Terminal but Not ER Lumen Region
The ER Lumen and C-terminus Sequences of NS2 Involved in Binding to PLA1A
NS5A Interacts with PLA1A through its Anchor Helix and DI Regions
PLA1A Stabilizes the NS2-E2 and NS2-NS5A Complex
Colocalization of PLA1A with E2, NS2 and NS5A
-
Previously, PLA1A was shown to interact with the HCV E2, NS2 and NS5A proteins and facilitate NS2-E2 and NS2-NS5A complex formation during HCV assembly (Guo et al. 2015). In this study, we revealed that HCV infection could up-regulate the PLA1A expression in vivo. The key domains of E2, NS2 and NS5A involved in PLA1A interaction to their lumenal domains and membranous parts were mapped, through which they form oligomeric protein complexes to participate in HCV assembly.
HCV assembly requires complex protein-protein interactions between HCV proteins and host factors. It has been generally accepted that the Core-driven LD and the replication complex (RC) are brought together through the interaction of Core and NS5A, which is very important at an early step of HCV assembly. NS2 protein is required for virus assembly as a scaffold recruiting viral envelope proteins to the assembly sites in close proximity to LD. The E1-E2-NS2-p7 complex migrates to a position close to the RC through the interaction between NS2 and NS5A. The argument is that the interaction between NS2 and NS5A was thought to be a transient or unstable interaction. This study provides a detailed characterization of the interaction between PLA1A and NS2, E2 or NS5A. PLA1A played an essential role as a bridge in HCV assembly by recruiting the NS2 complex together with E1-E2 glycoprotein to core-containing LDs via the interaction with NS2 and NS5A (Fig. 10).
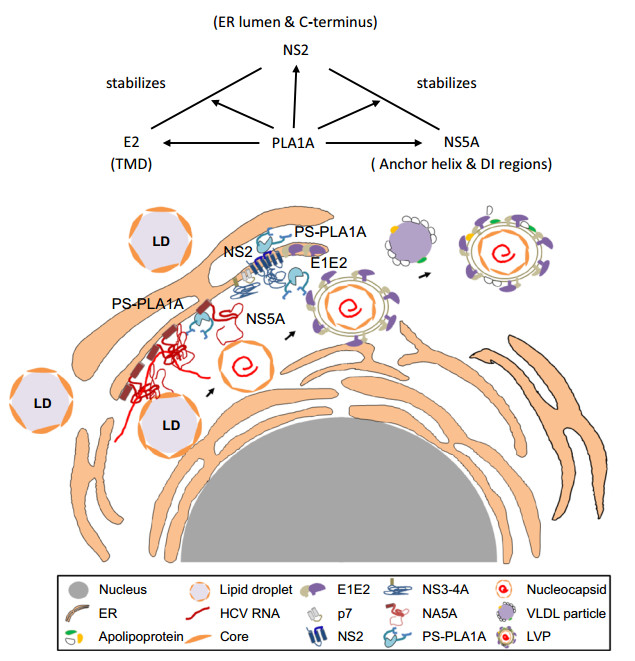
Figure 10. Working model of PLA1A involvement in HCV assembly through NS2 complex formation. PLA1A affects HCV assembly by stabilization the NS2/NS5A complex. Furthermore, PLA1A could interact with E2 and NS2, influencing the envelopment of HCV.
NS2 interaction protein was reported to mediate NS2- E2 complex formation, contributing to the HCV assembly process. It had been reported that NS2 is not essential for RNA replication; however, NS2 could bring together the viral E1-E2 glycoprotein complex, p7, and the NS3-4A enzyme complex. The interaction between NS2 and E1-E2 envelope protein has been shown using biochemistry and genetic data. A similar function for PLA1A in the NS2-E2 interaction was found in this research because PLA1A interacts with both E2 and NS2. Meanwhile, the interactions of PLA1A with NS2 or E2 were time-independent, but the interaction with NS5A was time-dependent. PLA1A could contribute to NS2-E2 and NS2-NS5A complex formation or stabilize the complex, demonstrating the functional importance of PLA1A in HCV assembly.
PLA1A specifically acts on phosphatidylserine (PS) and 1-acyl-2-lysophosphatidylserine (lyso-PS) to hydrolyze fatty acids at the sn-1 position of these phospholipids. We had proved PLA1A could be upregulated in vivo. Thus, PLA1A may also contribute to HCV particle production through the cleavage products of fatty acids such as arachidonic acid. As a secreted enzyme, PLA1A may also hydrolyze phosphatidylserine on the cell surface and produce lyso-PS, as HCV was previously shown to induce apoptosis of infected cells. lyso-PS had been described as a potent activator of histamine release from mast cells, as a growth inhibitor of T cells, and as a chemotactic substance for fibroblasts and tumor cells. So PLA1A makes a tremendous contribution to HCV lifecycle and pathopoiesia.
In summary, as a bridge, PLA1A, was identified as a participant in the HCV assembly process. Data from this study demonstrate detailed domains of PLA1A, NS2, NS5A and E2 involved in the interaction. These results provide clues for understanding the details of the molecular mechanism of assembly and formation of infectious HCV particles.
-
The authors appreciate Prof. Frank Chisari (The Scripps Research Institute, California) for the provision of African green monkey kidney epithelial Vero cells and the derivative cell lines, Huh-7.5.1. We are also thankful to Stanley M. Lemon (University of Texas Medical Branch, Galveston) for the provision of subgenomic replicon used in this work. We are grateful to Dr. Zongqiang Cui for the mCherry-based BiFC assay and Dr. Ding Gao for technical support in confocal microscopy. Thanks for the core facilities center of Wuhan institute of virology for technical help. This work was supported by grants of the National Basic Research Priorities Program of China (2015CB554304).
-
The study was designed by JC and QY. QY and JC carried out the experiments; QY, MG, YZ, XH, YW, CW, MY, RP, XC, JC analyzed data. JC and QY wrote the paper. XC checked and finalized the manuscript. All authors read and approved the final manuscript.
-
The authors declare that they have no conflict of interest.
-
All patients, their relatives, or both provided written informed consent for the use of their clinical and pathological information for research purposes, as well as its storage in the hospital's database. The Ethics Committee of the First Hospital of Jilin University and Tongji Hospital, Tongji Medical College of Huazhong University of Science and Technolog approved the methods and experimental protocols used in the present study, which were performed in accordance with the ethical standards of our institutional research committee and the tenets of the 1964 Declaration of Helsinki and its amendments, or comparable ethical standards.

 DownLoad:
DownLoad: