












-
Dear Editor,
Since the first outbreak in Brazil in 2015, Seneca Valley virus (SVV) associated with porcine idiopathic vesicular disease, has shown increasing geographic distribution. Cases of SVV have been reported from several countries including the United States (US), Colombia, Thailand, Canada, and China (Pasma et al. 2008; Zhang et al. 2015; Sun et al. 2017; Wu et al. 2017; Liu et al. 2018; Saeng-Chuto et al. 2018). SVV was first identified in the US in 2002 and is the onlymember of the genus Senecavirus in the family Picornaviridae (Hales et al. 2008; Leme et al. 2017). The SVV genome contains a large open reading frame (ORF) encoding a polyprotein, which is cleaved into various mature viral proteins including the leader protein (L), structural proteins(VP1, VP2, VP3, and VP4), and non-structural proteins (2A, 2B, 2C, 3A, 3B, 3C, and 3D) (Hales et al. 2008). Structural proteins bind to their receptor, anthrax toxin receptor 1, to mediate viral invasion and stimulate specific immunity of the host (Jayawardena et al. 2018). Non-structural proteins such as 3C suppress innate immune responses by degrading interferon regulatory factor 3 (IRF 3) and IRF7 and inhibit the activation of RIG-1, TBK1, and TRAF3 through deubiquitination (Xue et al. 2018). To date, more than 100 SVV strains have been identified. Until the emergence of HeN-1/2018, no recombinant SVV strain had been detected in China. The HeN-1/2018 strain is originated from USA/IA44952/2015 and USA/INPurdue-4885/2015 strains and its shortest recombinant fragment comprises a region of VP2, VP3 (partial), and VP4 (partial) genes (Wang et al. 2018). Here, we report two newly identifiedrecombinant SVV strains, CH-GDFS-2018 and CHGDJY-2018, which are isolated from slaughtered pigs.
A total of 80 samples of porcine inguinal lymph nodes were obtained from local pig slaughterhouses in Guangdong Province. Forty were obtained from Jieyang city in May 2018 and the other 40 were sourced from Foshan city in November 2018. Virus isolation, RNA extraction and genomic sequence amplification were performed according to our previous report (Liu et al. 2018). Two SVV recombinant strains, CHGDFS-2018 and CH-GDJY-2018 were successfully isolated from the samples. The complete genomic sequences were determined and their GenBank accession Nos. are MK284514 and MK284515 for CH-GDFS-2018 and CH-GDJY-2018, respectively. A phylogenetic tree was constructed using MEGA version 6.0 based on the neighbor-joining method with 1000 bootstrap replicates to provide confidence values for the clustering results. The genomes of CH-GDFS-2018 and CH-GDJY-2018 are homologous with 97.1% nucleotide and 98.0% amino acid identities. Sequence alignment of CHGDFS-2018 with all SVV sequences available in GenBank revealed that CH-GDFS-2018 shared the highest complete genomic identity with KS15-01 and US-15-39812IA (98.4%). Moreover, it shared 98.3% identity with USA/IA39812/2015-P1 and 98.2% identity with USA/GBI29/2015, SD15-26, and USA/IA46008/2015-Passage 1. CH-GDJY-2018 shared the highest identity with SVA/CHN/07/2017 (98.8%). Furthermore, it shared 98.7% identity with GD06/2017 and SVA/ CHN/08/2017, and 98.6% identity with AH01-CH-2016 and SVA/CHN/09/2017. Phylogenetic analysis showed that CHGDFS-2018 and CH-GDJY-2018 were clustered together with the strains isolated in China in 2017 and these all belonged to the US SVV branch (Fig. 1A). These results revealed that the SVV strains circulating in China are genetically diverse and complex.
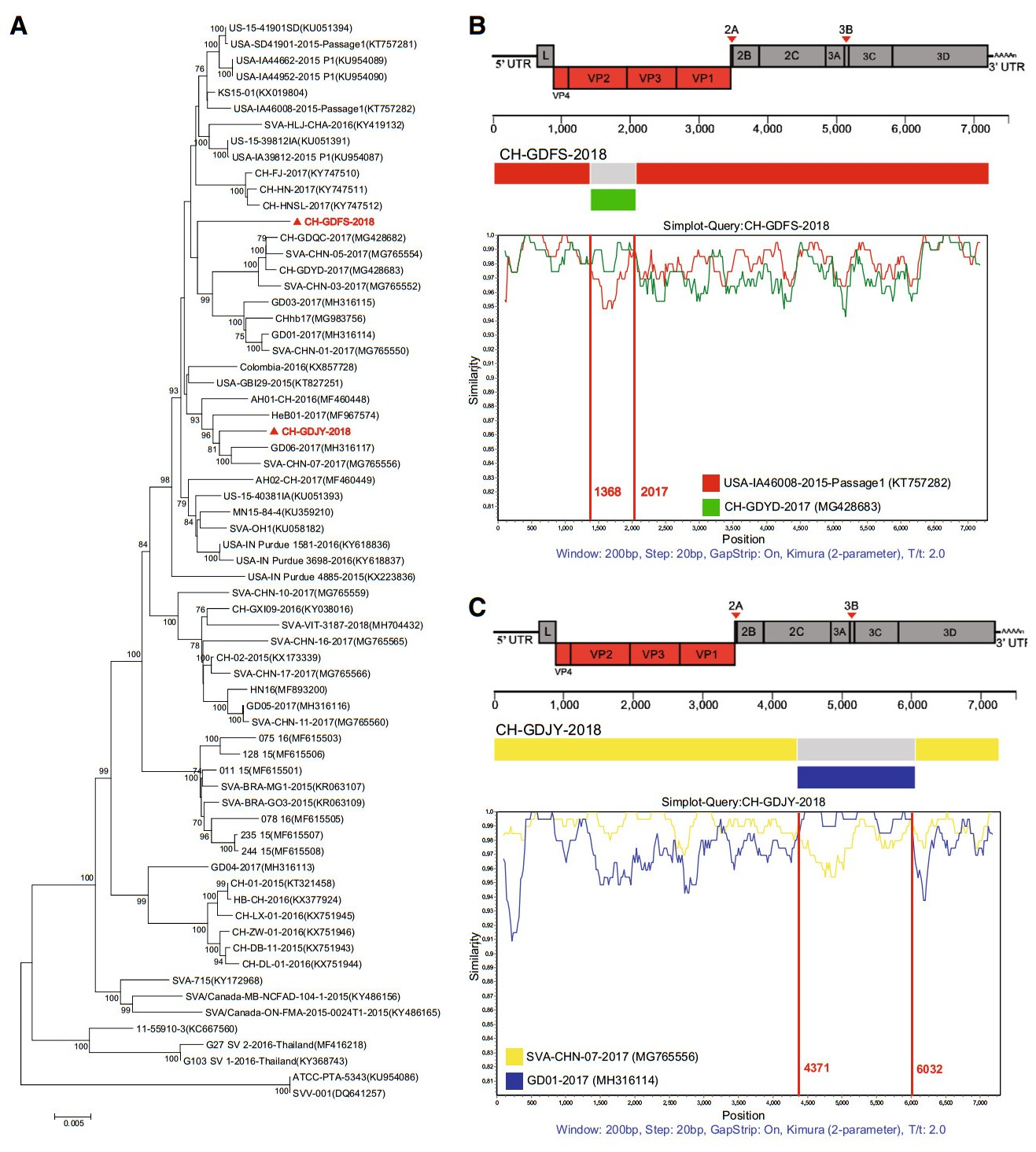
Figure 1. Phylogenetic analysis and recombination analyses of genomic sequences of two newly identified SVV strains, CH-GDFS-2018 and CH-GDJY-2018. A Phylogenetic analysis of the two strains and other SVV strains available in GenBank. The phylogenetic tree was constructed using the MEGA 6.0 software and the Tamura-Nei model with 1000 bootstrap replicates. Strains isolated in this study are red colored and highlighted with solid triangles. B Recombination analyses of CH-GDFS-2018. C Recombination analyses of CHGDJY-2018. Reference strains, USA-IA46008-2015-Passage1 (red), CH-GDYD-2017 (green), SVA-CHN-07-2017 (yellow), and GD01-2017 (blue), were used as putative parental strains. The X-axis indicates the location of the query sequence, and the Y-axis indicates the percentage of identity.
To validate recombinations in these strains, RDP4 software analysis was performed using different algorithms such as RDP, Bootscan, MaxChi, GENECONV, Chimaera, SiScan, and 3Seq (Martin et al. 2015). This analysis revealed that both CH-GDFS-2018 and CH-GDJY-2018 had undergone recombination events (P < 0.01 and recombinant score > 0.6). The results indicated that CH-GDFS-2018 was likely derived from USA-IA46008/2015-Passage 1, which was isolated in the US in 2015, and CH-GDYD-2017, which was identified in Guangdong Province in 2017. CH-GDJY-2018 seems to be derived from SVA-CHN-07-2017 and GD01-2017, which were epidemic field strains isolated in Guangdong Province in 2017. SimPlot and Bootscan analysis further confirmed genetic recombination patterns among CH-GDFS-2018, USA-IA46008/2015-Passage 1, and CH-GDYD-2017, and among CH-GDJY-2018, SVACHN-07-2017, and GD01-2017. The results showed that the recombinant genome of CH-GDFS-2018 consisted of two fragments from USA-IA46008/2015-Passage 1 (regions 1-1367 nt and 2018-7267 nt) and one fragment from CHGDYD-2017 (regions 1368–2017 nt) (Fig. 1B). Similarly, the recombinant genome of CH-GDJY-2018 consisted of two fragments from SVA-CHN-07-2017 (regions 1–4370 nt and 6033–7271 nt) and one fragment from GD01-2017 (regions 4371–6032 nt) (Fig. 1C). In the genome of CH-GDFS-2018, the shortest recombinant fragment comprised the region of VP2 (partial) and VP3 (partial) genes, while in CHGDJY-2018, the shortest recombinant fragment comprised the region of 2C (partial), 3A, 3B, 3C and 3D (partial) genes (Fig. 1B, 1C).
Guangdong is a major pig-rearing province of China. The first SVV infection case was reported here in 2015 (Wu et al. 2017) and infection reemerged here later in 2016 (Zhao et al. 2017) and 2017 (Chen et al. 2018; Liu et al. 2018). Surveillance of SVV transmission in Guangdong Province revealed that different branches of strains co-exist in swine herds, and this provided a potential platform for recombination to occur. Considering the diversity of epidemic field SVV strains in Guangdong Province and the fact that China imported a large number of breeding pigs from the US, it is possible that CH-GDFS-2018 and CH-GDJY-2018 are recombinant SVV strains. HeN-1/2018, which was the first recombinant SVV strain reported in China, was isolated from sick pigs. Its shortest recombinant fragment mainly comprised structural proteins (Wang et al. 2018). Contrastingly, CH-GDFS-2018 and CH-GDJY-2018 were isolated from slaughtered pigs, which had recovered from the vesicular disease, and the recombinant fragments of these strains comprised both structural and non-structural protein regions. Additionally, CH-GDFS-2018 and CH-GDJY-2018 were a result of recombination between local or domestic epidemic strains and foreign strains. Considering this adaptation of the virus to host, the occurrence of natural recombination is of clinical importance.
In summary, two novel recombinant SVV strains, CHGDFS-2018 and CH-GDJY-2018, were isolated and identified from the pigs in slaughterhouses in Guangdong Province. The significance of recombination in different regions of SVV genome needs further research for better understanding this novel pathogen. The findings of this study may help in developing better prevention strategies against SVV outbreaks.
HTML
-
This work was supported by the Key Research and Development Program of Guangdong Province (2019B020218004).
-
The authors declare that they have no conflict of interest.
-
All institutional and national guidelines for the care and use of animals were followed.

 DownLoad:
DownLoad: