














-
Dear Editor,
Hepatitis B virus (HBV) infection remains a major cause of liver diseases worldwide, which affects approximately 250 million people globally (Yuen et al. 2018). Individuals with chronic HBV infection are at high risk of developing liver cirrhosis, and ultimately hepatocellular carcinoma (HCC). HBV is a Hepadnavirus DNA virus, which specifically infects and efficiently replicates in hepatocytes. It has been proposed for decades that HBV replicates preferentially in non-dividing quiescent hepatocytes (Ozer et al. 1996), therefore remarkably influenced by cell growth and proliferation status. In line with this, a recent study by Carla Eller et al. (2020) reported that by utilizing a genome-wide gain-of-function screen, CDKN2C, the inhibitor of cyclin D (CDK4/6), was identified as an HBV host factor favoring viral replication via cell cycle arrest. In their study, cell cycle arrest is essential for generating a prerequisite cellular environment for CDKN2C to enhance HBV replication. Nevertheless, given the fact that the stealth HBV replicates most efficiently in the quiescent hepatocytes in the immune tolerance phase, it remains to be validated whether CDKN2C virtually serves as an HBV host factor.
To reevaluate the role of CDKN2C in HBV replication in patients of chronic hepatitis B (CHB), the relationship between CDKN2C expression and HBV load in CHB patients was analyzed by use of the data of 90 CHB patients in GSE83148 dataset (Zhou et al. 2017), yet no significant difference of CDKN2C expression between patients with HBV DNA load < lg106 IU/mL and those with ≥ lg106 IU/mL (Fig. 1A) was observed. To consolidate our finding, GSE84044 (Wang et al. 2017), the liver transcriptome dataset from another 124 patients with HBVrelated liver fibrosis was also analyzed. Either, no correlation between hepatic CDKN2C expression levels and serum HBV DNA load was observed (r = 0.115, P = 0.255) (Fig. 1B). Next, a third dataset of GSE94660 (Yoo et al. 2017) composing of RNA-seq data from 21 HBV infection-related HCC patients was analyzed. Similarly, liver CDKN2C expression showed no correlation with HBV RNA levels in the malignant hepatocellular carcinoma tissues either (r = 0.367, P = 0.107) (Fig. 1C).
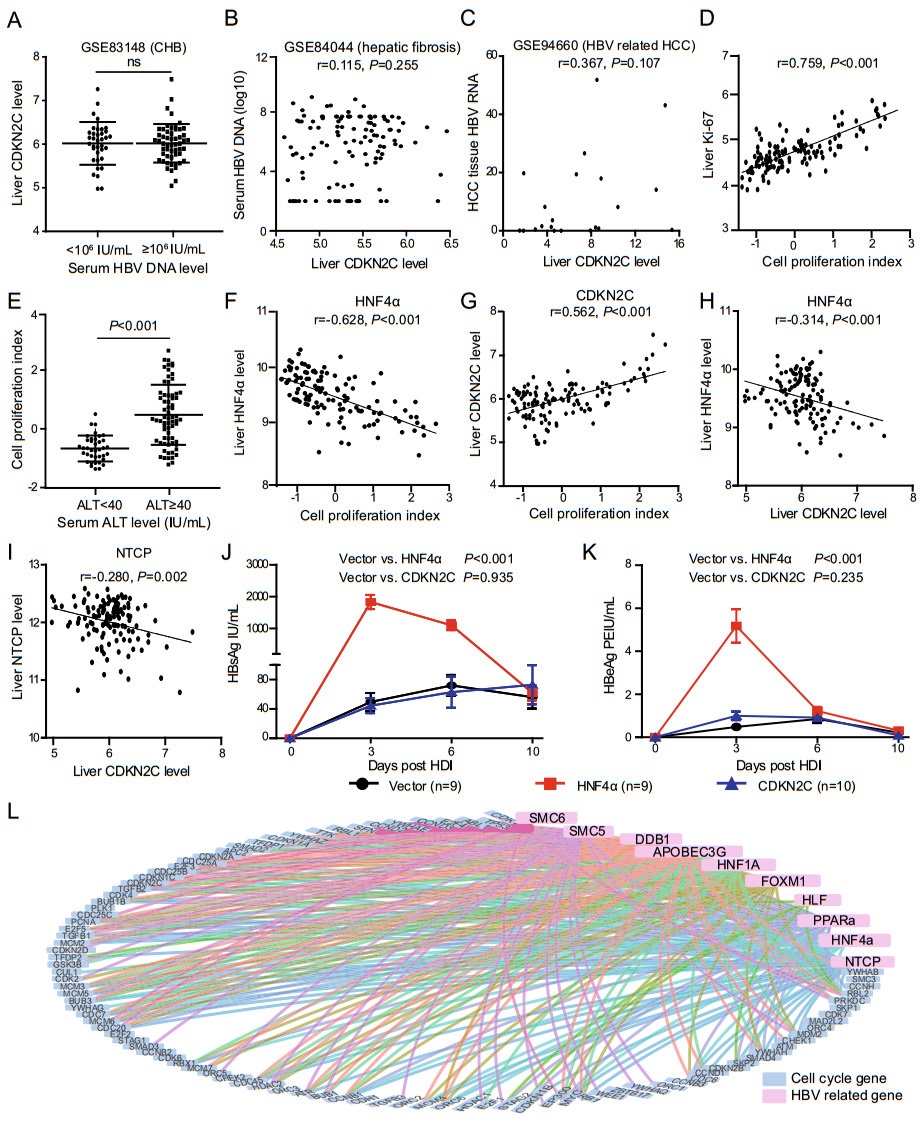
Figure 1. CDKN2C is not an HBV favoring host factor. A CDKN2C expression level in CHB patients with serum HBV DNA < lg106 IU/ mL and those with ≥ lg106 IU/mL in GSE83148 dataset. B Relationship between liver CDKN2C expression level and serum HBV DNA for patients with HBV-related liver fibrosis in GSE84044 dataset. C Correlation between CDKN2C expression level (FPKM) and liver HBV RNA level for HCC tissues in GSE94660 dataset. D Correlation between liver cell proliferation index and expression of Ki-67 in GSE83148 dataset. E Comparison of liver cell proliferation index in patients with chronic hepatitis B with different serum ALT levels in GSE83148 dataset. F, G Correlation between liver cell proliferation index and expression of HNF4α and CDKN2C in GSE83148 dataset. H, I Correlation between liver CDKN2C level and HNF4α, and NTCP expression level in GSE83148 dataset. J, K Serum HBsAg and HBeAg levels at different time points after the mouse tail vein hydrodynamic injection. L Analysis of expression correlation between cell cycle related genes and HBV-dependent host genes. Blue dots represent cell cycle genes and pink dots represent HBVdependent host genes. The line between the two points indicates that the expression levels of the two genes are highly correlated with the cut-off values |r| > 0.4 and fdr < 0.05.
It has been reported that the expression of CDKN2C was markedly upregulated when cells reach G1/S transition and progress through the remainder of the cycle (Hirai et al. 1995). Increased expression of CDKN2C has also been shown to be coordinated with the terminal differentiation of adipocytes (Morrison and Farmer 1999). To explore the relationship between CDKN2C expression level and hepatocyte proliferation status in CHB patients, we first developed a cell proliferating index model through factor analysis, utilizing the gene expression status of 92 out of the 116 KEGG annotated cell cycle related genes (ko04110, http://www.kegg.jp) available in GSE83148 dataset. This novel index could reflect the proliferation status of hepatocytes with good performance, as it was well positively correlated with Ki-67 levels (r = 0.759, P < 0.001) in the liver tissues of these CHB patients (Fig. 1D). Intriguingly, the cell proliferating index was significantly higher in patients with higher serum ALT levels than those with normal levels (UNL, 40 IU/mL) (Fig. 1E), indicating that necro-inflammatory injury causes hepatocyte compensatory proliferation. As expected, the score of cell proliferation index is significantly negatively correlated with HBV-dependent host gene HNF4α (r = -0.628, P < 0.001) (Fig. 1F), in consistent with the previous report (Hanse et al. 2012). However, contrary to Eller et al.'s report (Eller et al. 2020), the expression of CDKN2C was found significantly positively correlated with the cell proliferation index (r = 0.562, P < 0.001) in the liver tissue specimens derived from CHB patients (Fig. 1G). As mentioned above, CDKN2C acts as an inhibitor of CDK4/6 to block the cell cycle transition from G1 to S phase. Based on the fact that the expression of CDKN2C in the liver tissues of CHB patients is positively correlated with the proliferation index, we rationally inferred that the elevation of CDKN2C expression would be a secondary response to the transition of parenchymal hepatic cells from quiescence into rapid proliferation status, attempting to fulfill its function as a ''brake'' on hepatocytes' proliferation in CHB patients.
Since several studies have reported the inhibition of HBV replication induced by cell proliferation (Allweiss et al. 2018; Yan et al. 2019), we investigated the correlation between CDKN2C expression and the expressions of a series of HBV-related host factor genes known to enhance HBV transcription in GSE83148 dataset of CHB patients. Contrary to Eller et al.'s report that CDKN2C expression could enhance HBV RNAs transcription via upregulation of HNF4A, HLF, and PPARα, a series of HBV-related host factor genes known to enhance HBV transcription (Eller et al. 2020), we found that the mRNA level of CDKN2C was negatively correlated with HNF4α (r = -0.314, P < 0.001), NTCP (r = -0.280, P = 0.002) (Fig. 1H, 1I), HLF (r = -0.209, P = 0.021), and PPARα (r = -0.317, P < 0.001) (Supplementary Fig. S1A and Supplementary Fig. S1B). Similar results were obtained as well in a cohort of HBV infection-related liver fibrosis patients (GSE84044) (Supplementary Fig. S1C–S1F). Since CDKN2C is not positively correlated with the expression of HBV-dependent genes in the liver tissues of CHB patients, it is rather unlikely for CDKN2C to enhance HBV replication via upregulating these HBV-related host factor genes. Because most of the liver cells are in a quiescent (G0) state, we assume that CDKN2C cannot play the role of inhibiting cell proliferation and promoting HBV replication in vivo. To verify this hypothesis, we injected the HNF4α or CDKN2C expressing plasmids together with the same amount of 1.2mer HBV plasmids respectively into mice through tail vein hydrodynamic injection (HI), and vector pcDNA3.1 was used as control. The mouse serum was collected at 3, 6, and 10 days after HI, and the levels of HBeAg and HBsAg in the serum were measured. As expected, the levels of HBeAg and HBsAg in the HNF4α group are more than 15 times higher than that of the control, but there is no significant difference between the CDKN2C group and control group (Fig. 1J, 1K). The above results indicate that CDKN2C cannot promote HBV replication in vivo. Thereafter, we further investigated the relationship between other known cell cycle related genes and HBV-dependent host genes (NTCP, HNF4α, HLF, PPARα, HNF-1, HNF-3, APOBEC3, DDB1, SMC5, and SMC6) (Leupin et al. 2005; Yan et al. 2012; Decorsière et al. 2016; Turton et al. 2020) by using the data in the GSE83148 dataset. We found that almost all 92 cell cycle genes are related to at least one known HBV-dependent gene (Fig. 1L), indicating that all these cell cycle regulating genes have the potential to impact the expression of HBV-dependent genes and thereby HBV replication.
In summary, we demonstrated here that elevated CDKN2C expression was positively correlated with the proliferation state of the parenchymal liver cells, suggesting that CDKN2C expression in necro-inflammatory liver tissues, even though markedly upregulated, was not enough to facilitate the expression of the HBV host factor genes since it failed to repress cell cycle progression. Likewise, no correlation between CDKN2C expression and HBV replication in HBV-infected patients owing to the absence of CDKN2C expression in hepatocytes dominated in quiescent status and animal experiments also confirmed that CDKN2C cannot promote HBV replication in vivo. In conclusion, it is neither CDKN2C nor other single cell cycle specific gene itself, but instead the cell cycle arrest induced by them promotes HBV replication in patients with HBV infection. Hence, it is improper to term these genes as HBV host factors.
HTML
-
This work was supported by the National S & T Major Project for Infectious Diseases of China (Nos. 2017ZX10202202, 2017ZX10202203) and Beijing Natural Science Foundation (7182079).
-
The authors declare that they have no conflict of interest.
-
The animal experiments were conducted in accordance with the Guide for the Care and Use of Laboratory Animals and were approved by the animal ethics committee of Peking University Health Science Center (Beijing, China).

 DownLoad:
DownLoad: