












HTML
-
Japanese encephalitis virus (JEV), like dengue virus (DENV), Zika virus (ZIKV), and West Nile virus (WNV), belongs to the genus Flavivirus and family Flaviviridae and is the pathogenic agent of Japanese encephalitis (JE), which mainly prevails in Asia and Southeast Asia, renders more than two billion people at risk, and thus is the most important viral encephalitis in the world (Turtle and Solomon 2018). Unlike DENV or ZIKV, JEV is much more neuroinvasive, and JEV invasion into brain usually results in severe encephalitis. The average fatality rate of JE is 20%, in children the fatality can be 30% (Amicizia et al. 2018). About 50% of JE patients develop permanent neurological sequelae. Despite the great disease burden of JE, we currently know little about JE pathogenesis and have no specific therapy for it (Turtle and Solomon 2018).
Axl, a key member of receptor tyrosine kinase TAM family (the other two members are Tyro3 and Mertk), plays a key role in immune and inflammatory homeostasis (Rothlin et al. 2007). Axl is universally expressed in multitudinous cells, such as macrophages and dendritic cells (Rothlin et al. 2015). Gas6 broadly existing in the humoral fluid is the natural ligand for Axl. Cells utilize Axl to identify and bind to phosphatidylserines on the surface of apoptotic cells via bridging by Gas6, which promotes the phagocytic clearance of apoptotic cells and averts inflammatory response (Seitz et al. 2007). Ligation of Axl to phosphatidylserines also activates Axl signaling pathway and promotes the expression of suppressor of cytokine signaling (SOCS) 1 and SOCS3, which suppress the production of many pro-inflammatory cytokines such as TNF-α, IL-1β, and IL-6 (Rothlin et al. 2007; Sun et al. 2010). Interestingly, many enveloped viruses also hold phosphatidylserines on their surface, these phosphatidylserines can bind to Axl expressed on surface of host cells and facilitate virus entry (Moller-Tank and Maury 2014). Flaviviruses including DENV, ZIKV, and WNV can exploit Axl to promote entry (Richard et al. 2017). However, the roles of Axl in flavivirus infection are far beyond a receptor. During ZIKV infection, besides acting as an entry receptor, Axl also attenuates the production of type Ⅰ interferon and inflammatory cytokines to favor viral replication (Meertens et al. 2017; Chen et al. 2018; Strange et al. 2019).
The roles of Axl in encephalitis have been less studied. Activation of Axl signaling attenuates neuroinflammation and reduces neurological deficits after ischemic stroke (Wu et al. 2018). Loss of Axl signaling causes extensive axonal damage, prolonged neuroinflammation, and less remyelination after cuprizone exposure (Ray et al. 2017). Weinger et al. found that Axl-/- mice manifest more spinal cord lesions with larger inflammatory cuffs, more demyelination, and more axonal damage than wild type mice during experimental autoimmune encephalomyelitis (Weinger et al. 2011). These findings suggest that Axl plays an antiinflammatory and neuroprotective role during various kinds of encephalitis. We have previously reported that during JEV infection, Axl deficient mice displayed significantly more body weight loss than control mice, which we speculate it is the result of the anti-inflammatory role of Axl. Notwithstanding, there is no research discussing the role of Axl in regulating viral encephalitis, let alone Japanese encephalitis. Whether Axl negatively regulates inflammatory response in brain during JEV infection remains unknown.
In this research, by using Axl deficient mice and littermate heterozygous control, we discovered that Axl deficiency accelerated JE progression, which was correlated with exacerbated brain lesions induced by JEV, including increased neuron death, enhanced inflammatory cytokines production, and extended cytotoxic immune cells infiltration. These results suggest that Axl may be an antiinflammatory factor during JEV infection, which provides a potential target for alleviating inflammatory damage of JE.
-
C6/36 cells were grown in RPMI 1640 medium (Gibco, USA) supplemented with 10% fetal bovine serum (FBS, PAN, Germany) and maintained at 28 ℃. Vero cells were grown in minimum essential medium (MEM, Gibco, USA) supplemented with 5% FBS and maintained at 37 ℃. JEV Beijing strain-1 (JEV) (Sheng et al. 2016) was propagated in C6/36 cells and titrated on Vero cells by plaque assay as previously reported (Wang et al. 2018).
-
Axl deficient (Axl-/-) mice were kind courtesy of Dr. DaiShu Han (Peking Union Medical College, Beijing, China) and originally developed by Dr. Greg Lemke (Salk Institute for Biological Studies, La Jolla, CA). Mice were raised under a specific pathogen-free animal facility at Capital Medical University, China. The F0 Axl-/- mice were generated from C57BL/6 J mice and were mated with F0 wild type (Axl+/+) C57BL/6 J mice to generate F1 heterozygous (Axl+/-) mice. The F0 Axl-/- mice were then mated with F1 Axl+/- mice to generate F2 Axl-/- mice (experimental group) and littermate F2 Axl+/- mice (control group). The littermate Axl+/- mice are ideal controls for Axl-/- mice due to same age, similar nutritional and living status, and close genetic background. The genotype of F2 mice were identified by PCR, and three primers were used: Wt: 5'-GCCGAGGTATAGTCTGTCACAG-3'; Mut: 5'-TTTGCCAAGTTCTAATTCCATC-3'; WtMut: 5'-AGAAGGGGTTAGATGAGGAC-3'. The sizes of PCR products are 350 bp for Axl+/+ mice, 350 and 200 bp for Axl+/- mice, and 200 bp for Axl-/- mice.
-
Four-week-old Axl+/- and Axl-/- mice were intraperitoneally (i.p.) injected with 104, 105, and 106 PFU of JEV in 200 μL of phosphate buffered saline (PBS), or just 200 μL of PBS in mock-treated mice.
-
Brains of Axl+/- and Axl-/- mice were collected at 7 days post infection (dpi). The brains were immersed in 4% paraformaldehyde (PFA in PBS) for 24 h, then stored in 75% ethanol for paraffin embedding and section. Paraffin embedded brains were sagittally sectioned into 5 μm slides, which were then subjected to standard H&E staining procedures. The statistical analysis of HE staining results was completed by using Image-pro Plus 6.0 and GraphPad Prism 7.00 software.
-
Brains of Axl+/- and Axl-/- mice were harvested at 7 dpi. The left brains were embedded in OCT (4583, SAKURA) for cryosection. The right brains were immersed in 4% PFA for paraffin embedding and section. OCT embedded brains were sagittally cryo-sectioned into 6 μm slides and were fixed with ice cold acetone. Paraffin embedded brains were sectioned into 5 μm slides. For IF staining, after membrane permeability with 0.5% Triton and blockage with 1% bovine serum albumin (BSA), the brain sections were incubated with anti-JEV mouse serum (self-made) and anti-NeuN (ab104224, Abcam), anti-GFAP (ab7260, Abcam), or anti-Iba1 (10, 904-1-AP, Proteintech) in 1:300 dilution at 4 ℃ overnight and followed by incubation with Alexa Fluor 488 labeled donkey anti-mouse IgG antibody (A21202, Thermo) and Alexa Fluor 594 labeled donkey anti-rabbit IgG antibody (A21207, Thermo) in 1:500 dilution at room temperature for 40 min. For IHC staining, a heat mediated antigen retrieval was performed prior to primary antibody incubation with anti-CD4 antibody (ab183685, Abcam), anti-CD8 antibody (98941S, CST), and anti-SMI32 (SMI-32P, BioLegend), then an IHC kit (SNP-9001, Histostain) were used to visualize CD4, CD8, and SMI32 in brain tissues. For TUNEL staining, One Step TUNEL Apoptosis Assay Kit (C1086, Beyotime) was used according to the manufacture's instruction.
-
The mice were anesthetized and perfused with 60 mL of PBS at 7 days post infection. Brain tissue was isolated and the cerebral cortex and hippocampus were separated and immersed in digestive solution (0.25% trypsin and 0.02% EDTA in PBS) at 37 ℃ for 25 min with gentle shake followed by termination with equal volume of 10% FBS DMEM. After standing for 2 min, the supernatant was aspirated and filtered with 200 mesh filter followed by centrifugation at 1200 × g for 5 min. The cell pellet was resuspended with PBS and fixed with 32% paraformaldehyde to a final concentration of 4% at 37 ℃ for 10 min, followed by membrane permeability with 90% methanol on ice for 10 min. The cells were then blocked with 1% BSA at 37 ℃ for 10 min, followed by incubation with CD4 (553, 046, BD) and CD8 antibody (560, 182, BD) at room temperature for 1 h. The cells were then resuspended in PBS and subjected to flow cytometry by using Cytoflex system (Beckman).
-
Brains of Axl+/- and Axl-/- mice i.p. injected with 104 PFU of JEV were collected at 7 dpi and homogenized in 1 × cell lysis buffer (9803S, CST), followed by centrifugation at 12, 000 ×g for 10 min to collect supernatants. Resulting supernatants were subjected to a bead-based immunoassay (Aimplex) to measure the contents of a panel of cytokines (IL-1α, IL-1β, IL-2, IL-4, IL-6, IL-10, TNF-α, IFN-γ, CCL2, and CCL5).
-
The quantitative data were expressed as the mean ± SEM and all the statistical analyses were performed on GraphPad Prism 7.00 software. Student's t test or with Welch's correction and Mann-Whitney test were used to compare data between two groups. Difference was considered to be statistically significant when P < 0.05.
Cells and Virus
Mice
Animal Infection
Hematoxylin and Eosin (HE) Staining
Immunofluorescent (IF), Immunohistochemical (IHC), and TUNEL Staining
Flow Cytometry
Multiplex Immunoassay
Statistical Analysis
-
Axl-/- and Axl+/- (control) mice were i.p. injected with diverse dose of JEV (104, 105 and 106 PFU per mouse). Similar to the infection pattern in humans, in our mouse model, there were only two outcomes of JEV infection, namely inapparent and apparent infection (Fig. 1). Mice with inapparent infection showed no signs or loss of body weight, while mice with apparent infection manifested typical signs of JE and eventually died. About 6-7 days post infection (dpi), a fraction of mice, which positively correlated with infection dose, commenced to lose body weight and became sluggish (onset of encephalitis). As the disease progressed, these mice manifested ruffled fur, hunched back, limb paralysis successively, and finally died (Fig. 1). The time span from viral inoculation to the first observation of body weight loss was defined as the incubation period, and the time span from the first observation of body weight loss to death was defined as the disease duration. As expected, with the increase of infection dose, the incubation period was gradually shortened, but at all infection doses, the incubation period in Axl-/- and control mice was similar (Fig. 2A-2C). However, Axl-/- mice displayed shorter time of disease duration than Axl+/- mice (Fig. 2D-2F). These results suggest that Axl deficiency may accelerate disease progression in mice with apparent JE.
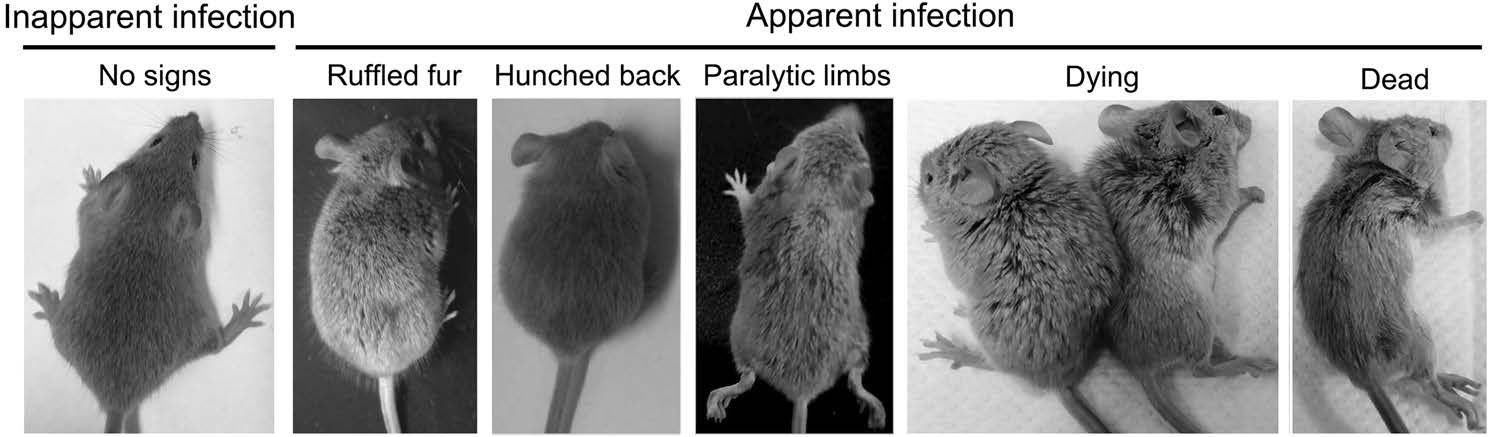
Figure 1. Signs of Japanese encephalitis virus (JEV) infection. Four-week-old Axl+/- and Axl-/- mice are intraperitoneally (i.p.) injected with 104 PFU of JEV. The infection patterns are classified into inapparent infection, where no signs of infection appear, and apparent infection, where the signs vary from ruffled fur, hunched back, paralytic limbs, dying, to death.
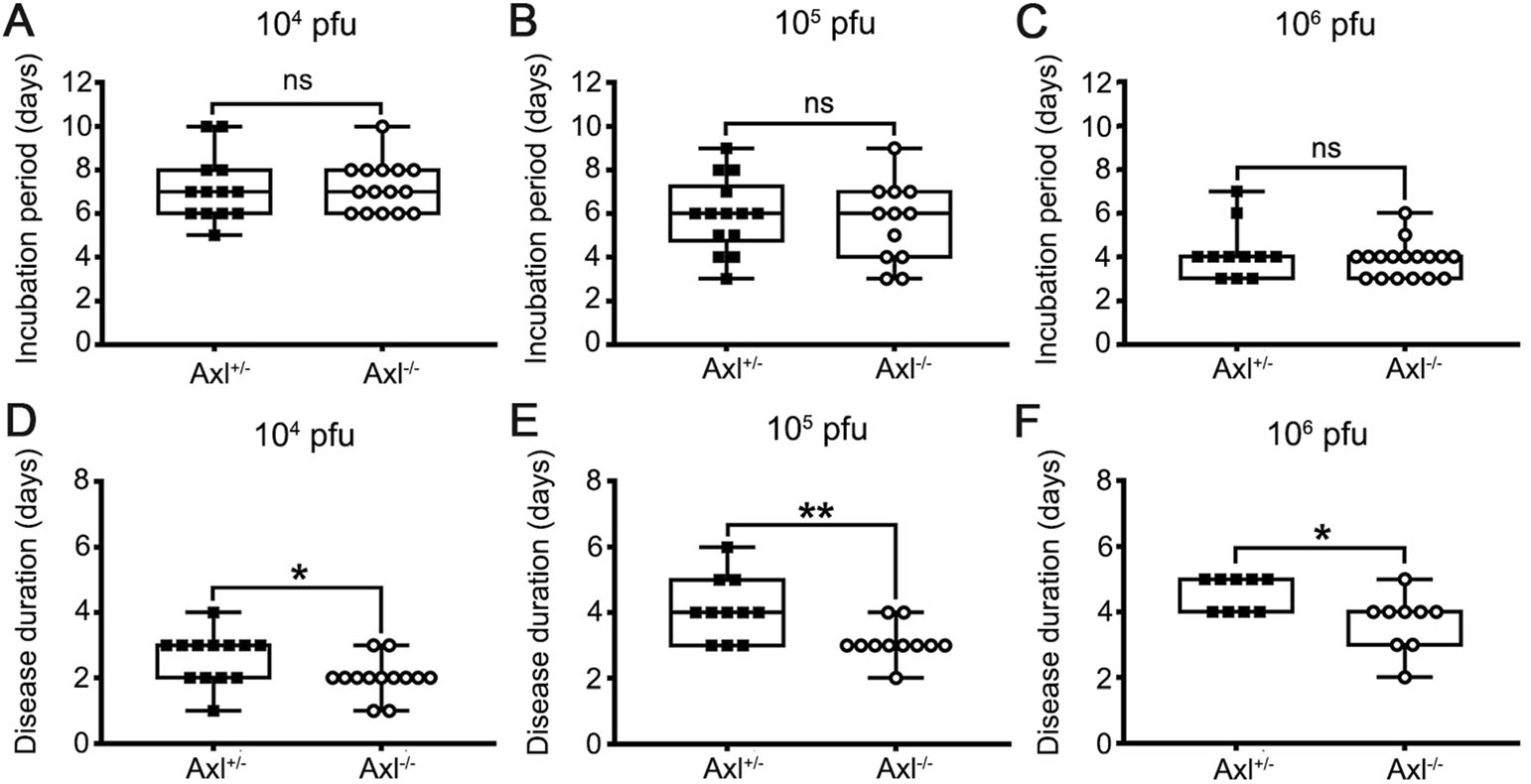
Figure 2. Incubation period and disease duration of Japanese encephalitis (JE). Four-week-old Axl+/- and Axl-/- mice are intraperitoneally injected with various dose of JEV. A-C Incubation period of Axl+/- and Axl-/- mice infected with 104 (A), 105 (B), and 106 (C) PFU of JEV. D-F Disease duration of Axl+/- and Axl-/- mice infected with 104 (D), 105 (E), and 106 (F) PFU of JEV. Data are expressed as the median + upper and lower quartiles, and each dot represents a mouse, * P < 0.05, as analyzed by Mann-Whitney test.
-
To test whether Axl deficiency accelerates JE progression by facilitating viral infection and/or exacerbating brain lesions induced by JEV, we examined viral infection and pathological changes in brains at 7 dpi using immunofluorescent (IF) staining and hematoxylin-eosin (HE) staining respectively. JEV antigens mainly distributed in cerebral cortex and hippocampus, and Axl-/- mice showed similar JEV distribution pattern in brain with control mice (Fig. 3). JEV mainly infected neurons (NeuN+), rather than astrocytes (GFAP+) or microglia (Iba1+) in brain, and Axl-/- mice displayed identical target cell tropism as control mice (Fig. 4). These results suggest that Axl is not an essential factor for JEV infection in brain. On the other hand, in both Axl-/- and control mice, conspicuous lesions in brains were observed, including neural cell loss (mainly in cerebral cortex and hippocampus), infiltration of inflammatory cells, angiectasis (vascular dilation), perivascular cuffing and disorganization of cerebral cortex architecture (Fig. 5A and 5B). Notably, Axl-/- mice showed much more neuron loss in cerebral cortex and hippocampus (Fig. 5A and 5C), more severe angiectasis in deeper cerebral cortex and cerebellum (Fig. 5A, 5B, and 5D), greater area of perivascular inflammatory cells infiltration than control mice (Fig. 5B and 5E). Neurofilaments (NF) are approximately 10 nm intermediate filaments found in neurons. They are a major component of the neuronal cytoskeleton, and function primarily to provide structural support for the axon and to regulate the axon diameter (Yuan et al. 2017). We observed the axonal damage by staining neurofilament and found that Axl-/- mice showed significantly reduced and thinned staining of neurofilaments in brain than Axl+/- mice during JE (Fig. 6). These results suggest that Axl deficiency enhances neuroinflammation and brain lesions during JE.
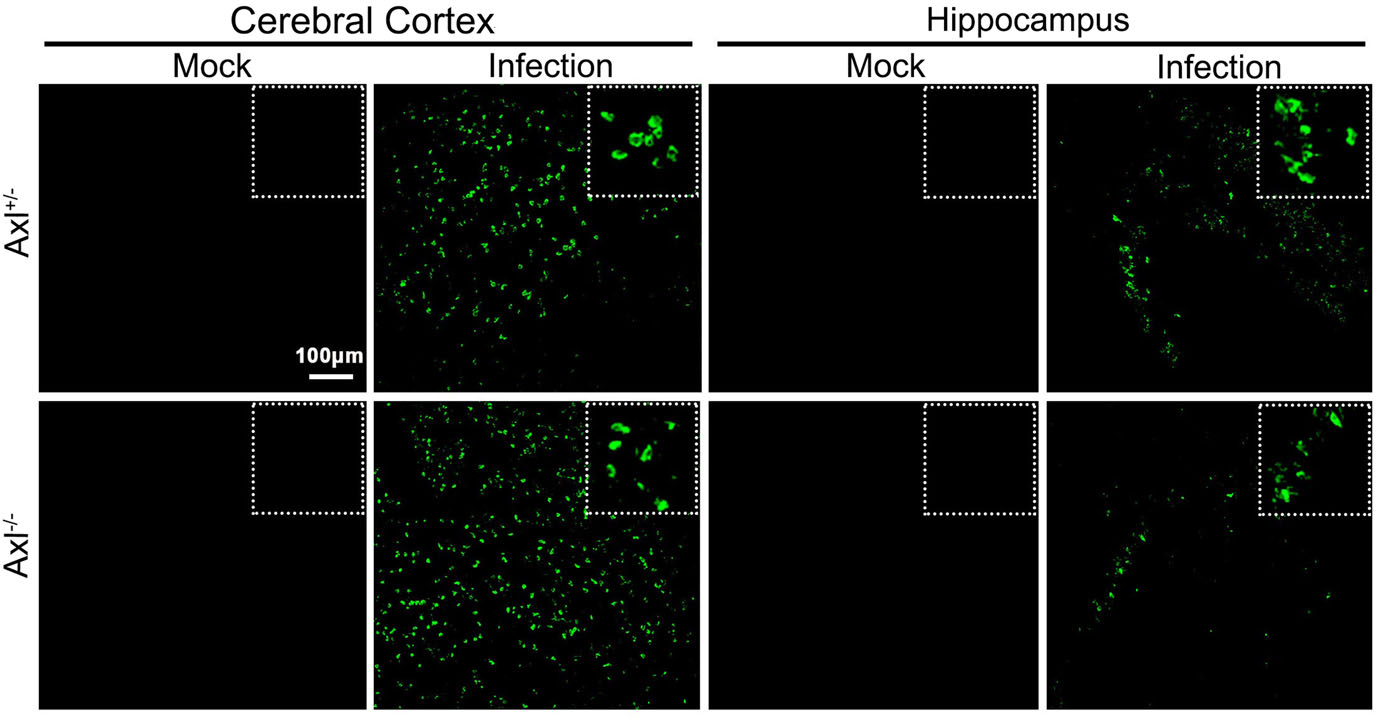
Figure 3. Distribution pattern of JEV in brain. Four-week-old Axl+/- and Axl-/- mice were intraperitoneally. injected with 104 PFU of JEV (infected) or PBS (mock). Brains were harvested at 7 dpi and subjected to IF staining of JEV antigens (green), scale bar = 100 μm.

Figure 4. Immunofluorescent costaining of JEV antigens and neural cell markers. Four-weekold Axl+/- and Axl-/- mice were intraperitoneally injected with 104 PFU of JEV. Brains were collected at 7 dpi and cryo-sectioned into 6 μm slides. JEV antigens (green) were costained with NeuN (a marker for neuron, red), GFAP (a marker for astrocyte, red) and Iba1 (a marker for microglia, red) in cerebral cortex. DAPI denotes nucleus, scale bar = 50 μm.
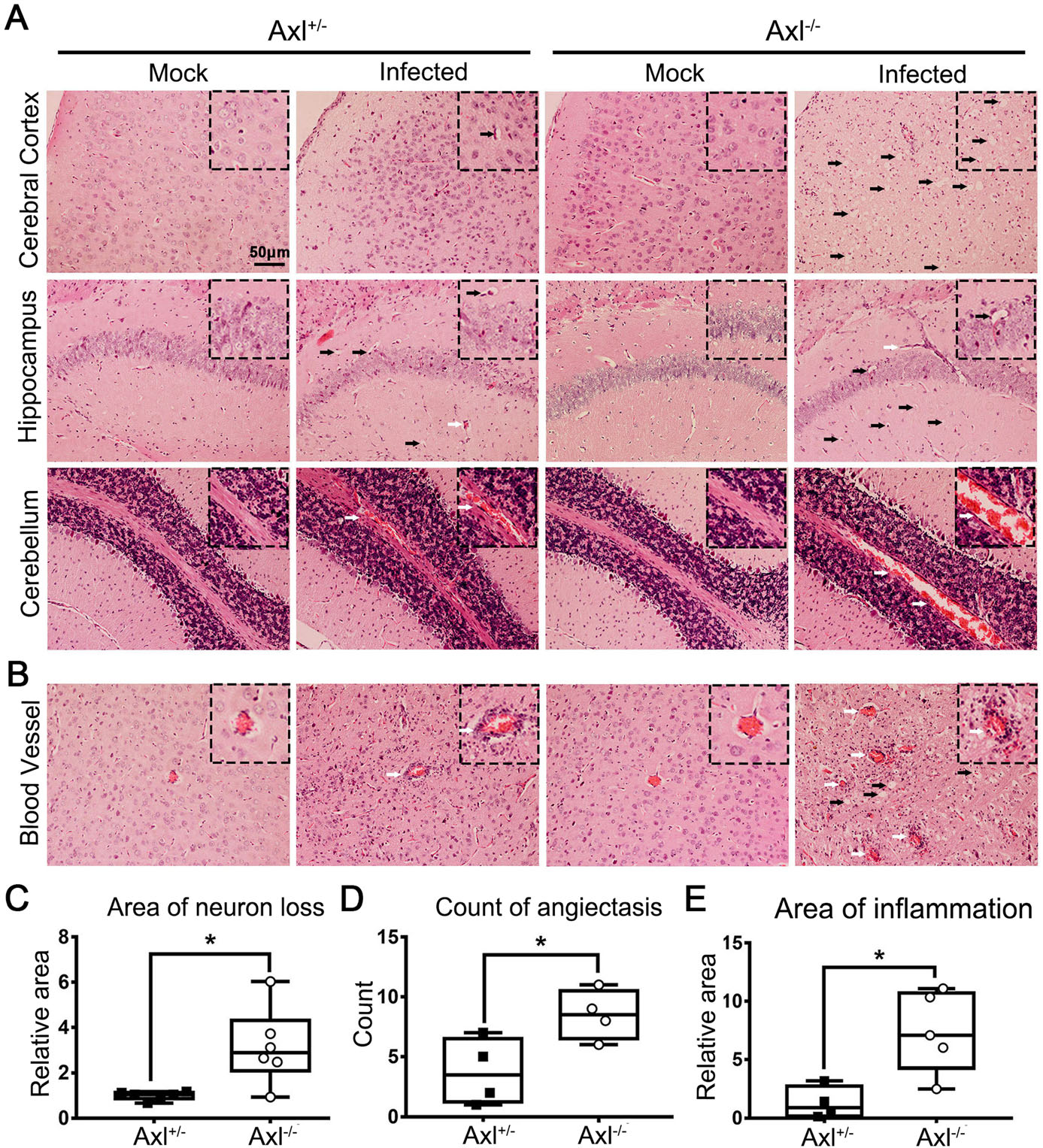
Figure 5. Pathological damage in brain during JE. Four-week-old Axl+/- and Axl-/- mice were intraperitoneally injected with 104 PFU of JEV (infected) or PBS (mock). Brains were collected at 7 dpi and sagittally sectioned into 5 lm slides. A, B HE image showing neuron death (black arrow) and angiectasis (white arrow) in brains of JE present mice at 7 dpi, scale bar = 50 μm. C-E Quantitative analysis of the area of neuron loss (C), count of angiectasis (D), and area of inflammatory cell infiltration (E). The area data of neuron loss and inflammatory cell infiltration were normalized to the average of JEV-infected Axl+/- mice. Data were expressed as the median + upper and lower quartiles, and each dot represents a mouse, * P < 0.05, as analyzed by Mann-Whitney test.

Figure 6. Immunohistochemical staining of neurofilament. Four-weekold Axl+/- and Axl-/- mice were intraperitoneally injected with 104 PFU of JEV (infected) or PBS (mock). Brains were collected at 7 dpi and sagittally sectioned into 5 μm slides. SMI32 was used to visualize neurofilaments in cerebral cortex. Black arrows denote neurofilaments; red arrows denote neuron death. This image is the representative of four Axl+/- and four Axl-/- mice.
We next study the effect of Axl on the apoptosis of JEVinfected neurons by using immunofluorescent staining of cleaved caspase 3 and TUNEL staining of damaged genome DNA. Although Axl-/- and Axl+/- mice showed similar expression of JEV in brain, Axl-/- mice showed significantly increased expression of cleaved caspase 3 (activated form) in brain (Fig. 7A). The apoptosis of neurons in Axl-/- mice was also more obvious than Axl+/- mice (Fig. 7B). These results suggest that Axl deficiency promotes the apoptosis of JEV-infected neurons.

Figure 7. The effect of Axl on the apoptosis of JEV-infected neurons. Four-week-old Axl+/- and Axl-/- mice were intraperitoneally injected with 104 PFU of JEV (infected) or PBS (mock). Brains were collected at 7 dpi and sectioned into 6 μm slides. A Immunofluorescent co-staining of JEV antigens and cleaved caspase 3, scale bar = 50 μm. B Immunofluorescent co-staining of NeuN and TUNEL (a marker for damaged DNA), scale bar = 50 μm. This image is the representative of four Axl+/- and four Axl-/- mice.
-
To further characterize the infiltration of inflammatory cells in brain, we performed immunohistochemical (IHC) staining of CD8+ T cells and CD4+ T cells. In brains of Axl-/- and control mice showing JE at 7 dpi, CD8+ T cells were widely distributed in whole brain, mainly in cerebral cortex, perivascular regions, hippocampus and cerebellum (Fig. 8A). Axl-/- mice showed greater number of CD8+ T cells in cerebral cortex (Fig. 8B), perivascular region (Fig. 8C), hippocampus (Fig. 8D), and cerebellum (Fig. 8E) than control mice. The infiltration of CD4+ T cells was also observed in brains (Fig. 8F). In comparison with the wide distribution of CD8+ T cells, CD4+ T cells were relatively sparse and mainly distributed in some superficial layers of cerebral cortex and some perivascular regions (Fig. 8F), and Axl-/- mice showed greater number of CD4+ T cells in cerebral cortex (Fig. 8G) and perivascular region (Fig. 8H) than control mice. We also detected the infiltration of T lymphocytes by using flow cytometry and found that consistent with the IHC results, Axl-/- mice displayed larger number of CD8+ T cells and CD4+ T cells in both cerebral cortex and hippocampus (Fig. 9A and 9B). The above data suggest that Axl deficiency enhances T lymphocytes infiltration in brain, which may promote brain damage during JE.
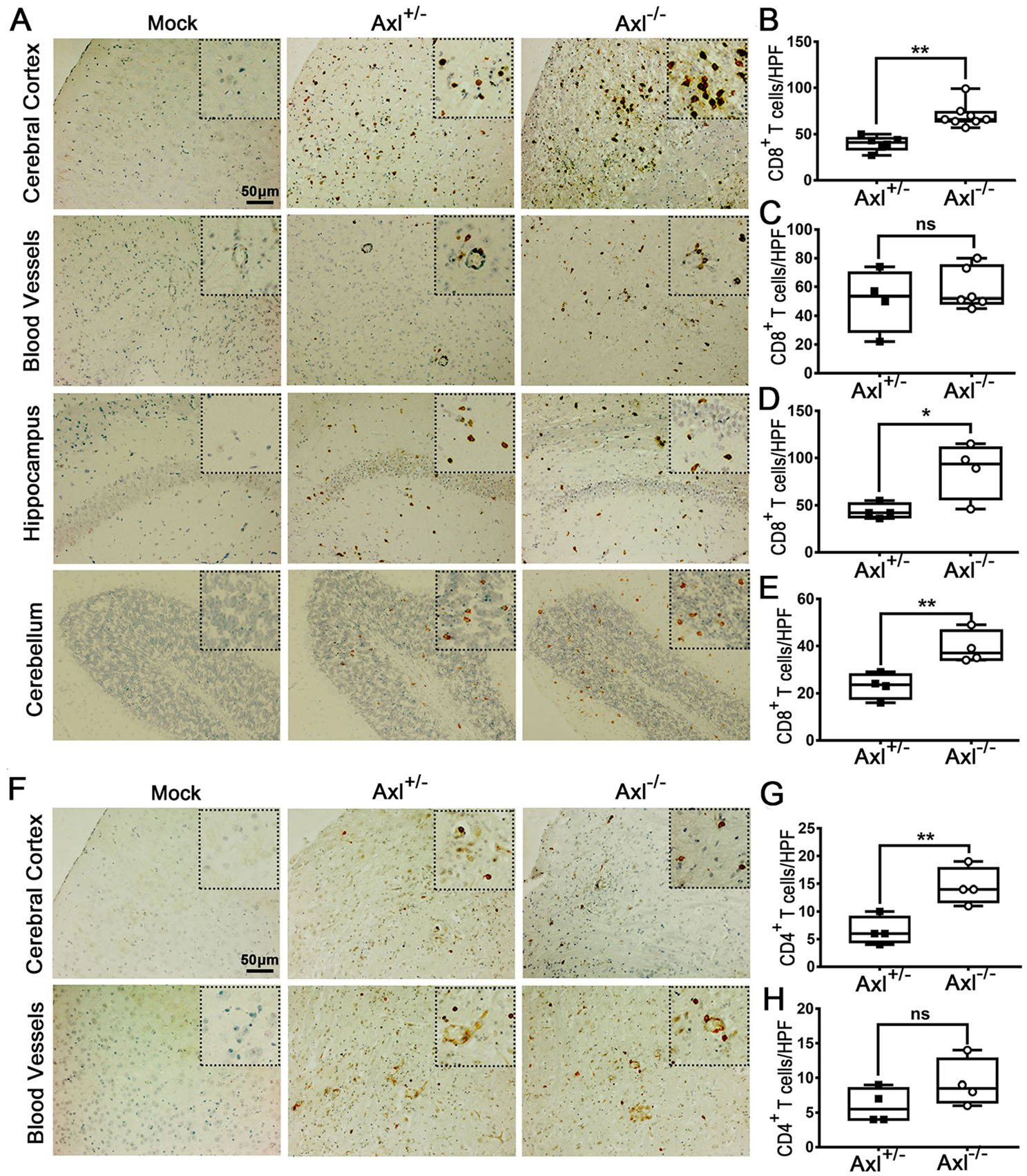
Figure 8. T lymphocytes infiltration in brain during JE. Four-week-old Axl+/- and Axl-/- mice were intraperitoneally injected with 104 PFU of JEV (infected) or PBS (mock). Brains were collected at 7 dpi and sectioned into 5 μm slides. CD8+ T cells and CD4+ T cells were demonstrated by IHC staining. A Representative IHC image depicting CD8+ T cell infiltration in brain at 7 dpi of JE present mice, scale bar = 50 μm. B-E Quantitative analysis of the number of CD8+ T cells per high power field (HPF) in cerebral cortex (B), perivascular region (C), hippocampus (D), and cerebellum (E). F Representative IHC image depicting CD4+ T cell infiltration in brain at 7 dpi of JE present mice, scale bar = 50 μm. G, H Quantitative analysis of the number of CD4+ T cells per HPF in cerebral cortex (G) and perivascular region (H). Data were expressed as the median + upper and lower quartiles, and each dot represents a mouse, *P < 0.05, **P < 0.01, and ns P > 0.05, as analyzed by Mann-Whitney test.
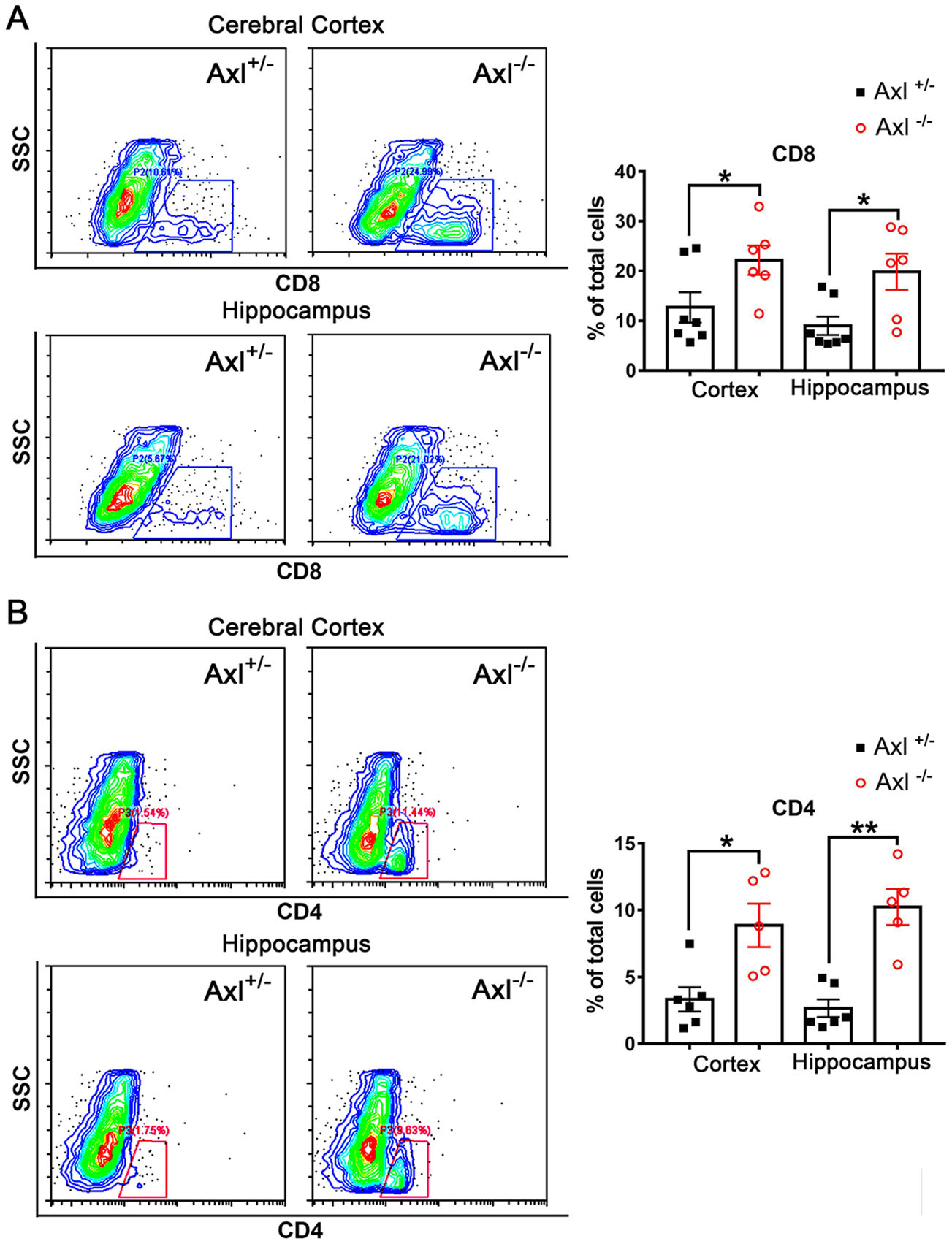
Figure 9. Detection of T lymphocytes in brain by flow cytometry. Percentage of CD8+ T cells (A) and CD4+ T cells (B) in cerebral cortex and hippocampus at 7 dpi. Each dot denotes a mouse, *P < 0.05, **P < 0.01, as analyzed by Student's t test.
-
We next measured a panel of cytokines (IL-1α, IL-1β, IL-2, IL-4, IL-6, IL-10, TNF-α, IFN-γ, CCL2, and CCL5) in brain at 7 dpi, which mediate inflammatory response. Under mock treatment, Axl-/- mice showed similar baseline levels of all the assayed cytokines with control mice (Fig. 10, mock). After infection, IL-2, IL-6, IFN-γ, CCL2, and CCL5 significantly elevated in brain, and Axl-/- mice showed much higher levels of these cytokines than control mice (Fig. 10, infected). These results suggest that Axl deficiency aggravates the production of pro-inflammatory cytokines in brain after JEV infection. These pro-inflammatory cytokines may work together with the infiltrated inflammatory cells to enhance neuroinflammation and deteriorate brain damage.
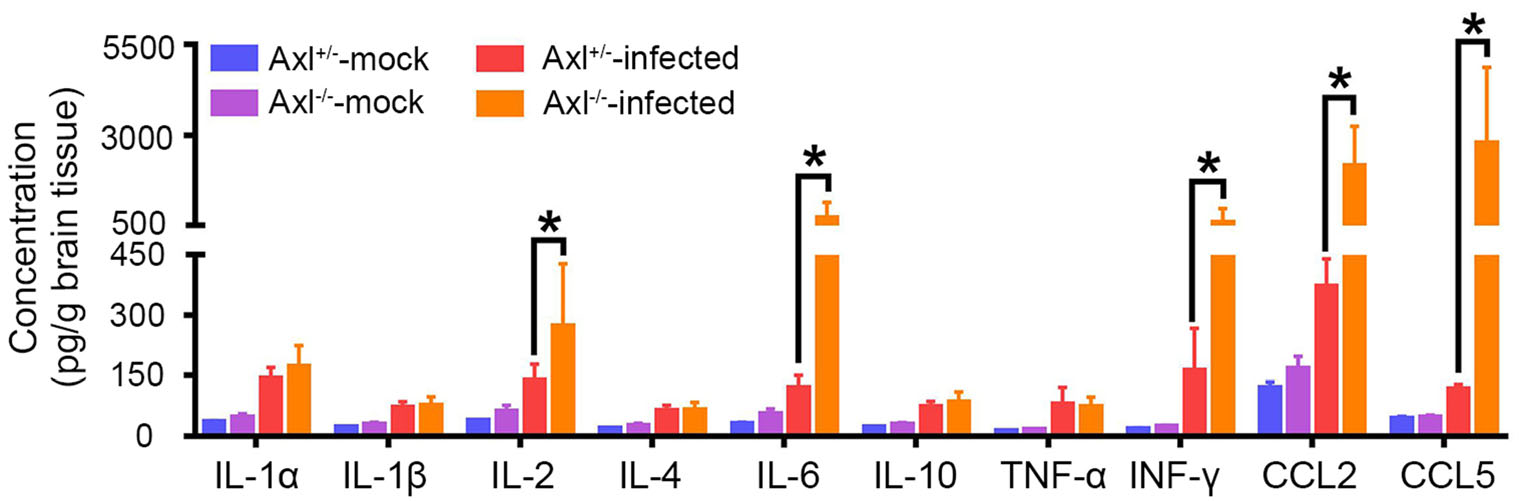
Figure 10. Inflammatory cytokines measurement in brain. Four-weekold Axl+/- and Axl-/- mice were i.p. injected with 104 PFU of JEV (infected) or PBS (mock). Brains were collected at 7 dpi and subjected to Aimplex immunoassay for cytokines measurement. Data are expressed as the mean ± SEM, n = 6 for each group, *P < 0.05, as analyzed by Student's t test.
Axl Deficiency Accelerates the Progression of Japanese Encephalitis (JE)
Axl Deficiency Exacerbates JE-Induced Brain Damage
Axl Deficiency Enhances T Lymphocytes Infiltration in Brain
Axl Deficiency Boosts Pro-Inflammatory Cytokines Production in Brain
-
JEV infection in brain causes a severe encephalitis called JE, which threatens people's health in 24 countries and is the most commonly diagnosed viral encephalitis in the world. Unfortunately, we have very limited countermeasures to specifically therapy JE, which is due to insufficient knowledge about JEV and JE pathogenesis. Axl has been reported to alleviate inflammatory damage through inhibiting production of a range of pro-inflammatory cytokines. However, its role in encephalitis has been less studied, let alone viral encephalitis. We have previously reported that during JEV infection, Axl deficient mice manifested more severe body weight loss than control mice, which we think is probably due to the loss of antiinflammatory effect of Axl. In this study, by using Axl-/- and control mice, we assessed the role of Axl in JE pathogenesis. We found Axl deficiency accelerated JE progression, which is associated with exacerbated brain lesions, characterized with increased neuron apoptosis, extended inflammatory cells infiltration, and enhanced production of pro-inflammatory cytokines. Our results thus reveal an anti-inflammatory and neuroprotective role of Axl in JE and suggest Axl may be a therapeutic target for JE.
Axl is a key member of TAM family, which has been reported to attenuate production of pro-inflammatory cytokines in multitudinous cells and tissues (Rothlin et al. 2007; Siemann et al. 2017). Activation of Axl signaling mitigates lung edema and alveolar inflammation in acute lung injury induced by ischemia-reperfusion by inhibiting production of pro-inflammatory cytokines including IL-1β, IL-6, and TNF-α (Peng et al. 2019). In a mouse model of autoimmune thyroiditis, stimulation of Axl signaling by Gas6 significantly reduces the incidence of thyroiditis, the infiltration of lymphocytes, and the production of pro-inflammatory cytokines (Sun et al. 2019). Axl also reduces severity of arthritis in a mouse model of arthritis (Waterborg et al. 2019). However, in some diseases, Axl plays contradictory role in regulating production of proinflammatory cytokines and chemokines. In a mouse model of glomerulonephritis, knockout or inhibition of Axl significantly reduces production of renal inflammatory cytokine and chemokine and improved kidney function (Zhen et al. 2018). Axl activation by Gas6 in pregnant rats significantly increases level of diverse pro-inflammatory cytokines in sera such as TNF-α, IFN-γ, IL-1α, IL-1β, and IL-6, and also increases placenta oxidative stress and apoptosis (Hirschi et al. 2019). Both Axl and its ligand Gas6 are abundantly expressed in the central nervous system, suggesting that Axl may have important function in brain (Shafit-Zagardo et al. 2018). However, up to now, the studies pertaining to the roles of Axl in encephalitis are very few, let alone viral encephalitis. Activation of Axl signaling alleviates neuroinflammation and neurological deficits after ischemic stroke (Wu et al. 2018). Axl knockout causes extensive axonal damage, prolonged neuroinflammation, and less remyelination after cuprizone induced encephalitis (Ray et al. 2017). Axl knockout leads to enhanced inflammation in the CNS and delayed removal of myelin debris during experimental autoimmune encephalomyelitis (Weinger et al. 2011). TAM triple knockout mice exhibit systemic autoimmune diseases, characterized by increased pro-inflammatory cytokine production, autoantibody deposition and autoreactive lymphocyte infiltration into brain (Li et al. 2013). In our own results, Axl deficiency increases neuron apoptosis and axonal damage, promotes production of several pro-inflammatory cytokines and chemokines, and facilitates infiltration of cytotoxic immunocytes, suggesting that Axl is an anti-inflammatory and neuroprotective factor during JE.
The molecular mechanism by which Axl regulates the inflammatory response in brain remains unclear and needs to be further studied. Notwithstanding, some investigations prove that activation of Axl signaling can inhibit Toll like receptors (TLRs) and TLRs-mediated cytokines production in human astrocytes, human glial cells, and human Sertoli cells via upregulating SOCS1 and SOCS3 (Rothlin et al. 2007; Meertens et al. 2017; Chen et al. 2018; Strange et al. 2019). This mechanism has been demonstrated to exist universally in various cell types, which may shed light on the understanding of the mechanism underlying Axl's antiinflammation function during JE.
In summary, our work revealed that Axl alleviates brain damage and delays encephalitis progression by inhibiting the generation of pro-inflammatory cytokines, impeding the infiltration of cytotoxic immune cells, and promoting the survival of neural cells. These results suggest that Axl is a neuroprotective factor against JE pathogenesis and it may act as a potential target for alleviating inflammatory damage of JE.
-
This work was supported by the National Natural Science Foundation of China (81671971, 81871641, 81972979, U1902210 and U1602223), the Scientific Research Plan of the Beijing Municipal Education Committee (KM201710025002), and the Key Project of Beijing Natural Science Foundation B (KZ201810025035), the Support Project of High-level Teachers in Beijing Municipal Universities in the Period of 13th Five-year Plan (IDHT20190510).
-
ZY Wang designed and performed most of the experiments, analyzed the data, wrote and reviewed the manuscript. ZD Zhen performed flow cytometry experiments and TUNEL staining. DY Fan maintained cells, viruses, and reagents. PG Wang and J An conceived the project, analyzed the data, and finalized the manuscript.
-
The authors declare that they have no conflict of interest.
-
All institutional and national guidelines for the care and use of laboratory animals were followed, and all the animal experiments were approved by the Experimental Animal Welfare and Animal Ethics Committee of Capital Medical University, Beijing, China (permission code: AEEI-2015-048; permission date: April 20, 2015).

 DownLoad:
DownLoad: