












HTML
-
Hepatitis B virus (HBV) is a hepatotropic, enveloped virus of the Hepadnaviridae family with a partial double-stranded relaxed circular DNA genome (rcDNA) (Bruss 2007). Upon infection, rcDNA is transported into the nucleus and converted into an episomal covalently closed circular DNA (cccDNA), which serves as the template to transcribe pre-genomic RNA (pgRNA) and other subgenomic viral RNAs. HBV genome encodes four transcripts corresponding to core/capsid protein (HBc), envelope protein (HBs), reverse transcriptase (Pol) and regulatory protein (HBx). Four promoters S1P, S2P, XP, and CP regulate and control the transcription of viral pre-genome RNA and proteins. Virus assembly begins with the formation of nucleocapsids by HBc, which are further enclosed by the large (LHBs), middle (MHBs), and small (SHBs) envelope proteins (Heermann et al. 1984).
Na+/K+ ATPase is an integral membrane protein responsible for establishing and maintaining the electro-chemical gradients of Na and K ions across the plasma membrane, which are essential for osmoregulation, sodium-coupled transport of a variety of organic and inorganic molecules, and electrical excitability of nerve and muscle (Kaplan 2002). Na+/K+ ATPase is composed of catalytic α-subunit and regulatory β-subunit that is required for proper trafficking, localization and functionality of the α-subunit. Three β-subunits (ATP1B1, ATP1B2, and ATP1B3) have been described in mammals (Clausen et al. 2017). Recent studies reported the involvement of ATPase family members in various virus infections. ATP1B1 was identified as a partner of human cytomegalovirus (HCMV) UL136 protein as well as M2 proteins of influenza A and B viruses (IAV and IBV) (Mi et al. 2010; Cui et al. 2011). ATP1A1 is critical for coronavirus, murine hepatitis virus, feline infectious peritonitis virus and vesicular stomatitis virus infection (Burkard et al. 2015). ATP1B3 was found to reduce BST-2-mediated restriction of human immunodeficiency virus type 1 (HIV-1) production in Hela cells (Nishitsuji et al. 2016). Inversely, ATP1B3 was shown to interact with the 3A protein of Enterovirus 71 (EV71) and inhibit EV71 replication by enhancing the production of type-I interferon (Lu et al. 2016). These findings suggest that ATP1B3 involves in RNA virus infection and replication, but there are relative few reports related to DNA virus like HBV until now.
We recently showed that ATP1B3 inhibits HBV replication via inducing NF-κB activation in HepG2 cells (Zheng et al. 2020). In this study, we further demonstrated that ATP1B3 inhibits HBV replication in HBV-producing cells HepG2.2.15 and HepAD38 cells, and determined that ATP1B3 has no effect on intracellular levels of HBV DNA and RNA. Due to the finding that ATP1B3 mutants which are defective in NF-κB activation still has the ability to inhibit HBV replication, we found that ATP1B3 can induce the degradation of the HBV envelope proteins through proteasomal pathway. Our works highlight ATP1B3 as a potential therapeutic target in HBV infection.
-
HepG2.2.15, HepAD38, Huh7, HEK293T, and Hela cells were maintained in Dulbecco's Modification of Eagle's Medium (DMEM; HyClone, Utah, USA) supplemented with 10% fetal bovine serum (FBS; GIBCO BRL, Grand Island, NY, USA), 1 mmol/L Na pyruvate, 100 μg/mL penicillin and 100 μg/mL streptomycin at 37 ℃ in a 5% CO2 incubator.
The following antibodies and reagents were used: human ATP1B3 (#ab137055, Abcam, Cambridge, UK), β-tubulin (#RM2003, Ray Antibody Biotech, Beijing, China), myc antibody (#05-724, Millipore, Billerica, Massachusetts, USA), anti-flag mouse monoclonal antibody (#F1804, Sigma, New York, USA), anti-HBs (Ad/Ay) antibody (horse polyclonal, #ab9193, Abcam), and anti-V5 monoclonal antibody (#R960-25, Invitrogen). AP-conjugated anti-rabbit IgG antibody and AP-conjugated anti-mouse IgG antibody were purchased from Jackson (#115-055-062, #115-055-045, Lancaster, Pennsylvania, USA). All general chemicals were purchased from Sigma, Selleck and TAKARA unless otherwise stated.
-
The expression constructs of ATP1B3-wild type, ATP1B3-∆NT, ATP1B3-∆TM, ATP1B3-∆CT (V5 tag), LHBs (myc tag), MHBs (myc tag), SHBs (flag tag), HBc (myc tag), ubiquitin, and HBV promoter fused luciferase reporter plasmids were generated by cloning the sequence of the coding region into a VR1012 expression vector. pCMV-HBV (genotype D), pHBV1.3 (genotype D) were kind gifts from Prof. Jianhua Li, School of Basic Medical Sciences, Fudan University. Ubiquitin (Ub), Ub-K48, Ub-K63, and Ub-KO plasmids were described previously (Li et al. 2020). Transfection with plasmids was performed using Lipofectamine 2000 according to the manufacturer's protocol. All transfection experiments were conducted with corresponding empty control.
Cells were transfected with siRNAs at a final concentration of 30–100 nmol/L using Lipofectamine RNAiMAX (Invitrogen) according to the manufacturer's protocol. At 72 h post-transfection, the cells were harvested to use for each assay. siATP1B3: 5′-UUUUGGAACCUCAUCGUUGAG-3′ and 5′-CAACGAUGAGGUUCCAAAAUA-3′.
-
The cultured mediums were examined for HBV surface antigen (HBsAg) with ELISA kits (Kehua, Shanghai, China). The samples were incubated in the 96-well microplates at 37 ℃ for 1 h, followed by addition of horseradish peroxidase-conjugated primary antibodies for 30 min, substrate for 10 min, and finally termination buffer. The microplate was quantified by a microplate reader (Bio-Rad, Hercules, CA, USA).
-
Detailed experimental procedures of HBV virions isolation were described in our previous report (Liu et al. 2020). Briefly, HBV virions were immunoprecipitated from culture supernatant by anti-HBs antibody conjugated to protein G beads (#11243233001; Roche) overnight at 4 ℃. In control groups, samples were incubated with beads only. For virion associated HBV DNA detection, the precipitate was digested with RQ1 RNase-Free DNase (#M6101, Promega, Madison, WI, United States) and Plasmid-safe DNase I (#E3101K; Epicentre Inc., San Diego, CA, United States) according to the manufacturer's protocol. The viral DNA was further extracted and detected by Southern blot and real time-PCR.
-
HBV DNA was extracted from the cells using a QIAamp mini kit (QIAGEN) according to the manufacturer's protocol. HBV total RNA was isolated from cell samples using TRIzol reagent (Invitrogen), and digested with DNase I (Life Technologies). The extracted DNA and extracted RNA was loaded on a 1.2% agarose gel in 1 × TAE buffer or on formaldehyde denaturing agarose gel, respectively, and then transferred onto a positively charged nylon membrane (Roche, Switzerland) using a vacuum blotter (Bio-Rad, USA). HBV DNA and RNA were hybridized and detected using a DIG Northern Starter Kit (Roche, Switzerland) according to the manufacturer's instructions.
-
Real-time PCR (for quantification of HBV DNA) and reverse transcription real-time PCR (for detecting HBV RNA) were performed essentially as described previously (Sommer et al. 2016; Iwamoto et al. 2019). The primer sets used in this study were 5′-GTTGCCCGTTTGTCCTCTAATTC-3′ and 5′-GGAGGGATACATAGAGGTTCCTTGA-3′ for HBV total DNA, 5′-GTGCACTTCGCTTCACCTCT-3′ and 5′-AGCTTGGAGGCTTGAACAGT-3′ for HBV total RNA.
-
Huh7 cells plated in 24-well plates were transfected with ATP1B3 expression plasmid along with four HBV promoters fused luciferase reporter plasmid. Two days later, cells were lysed, and firefly luciferase was examined with the Luciferase Reporter Assay System (Promega) according to the manufacturers' instructions.
-
10-cm dishes of HEK293T cells were transfected as indicated. 48 h post-transfection, cells were harvested and lysed with buffer containing a protease inhibitor cocktail (Roche, Indianapolis, IN). Cell lysates were incubated with anti-V5 or anti-myc antibody and protein G agarose beads (Life Technologies) at 4 ℃ overnight. After washing and elution, the eluted proteins were obtained by centrifugation, followed by western blot analysis.
-
The cells were harvested and boiled in 1 × loading buffer (0.08 mol/L Tris, pH 6.8, with 2.0% SDS, 10% glycerol, 0.1 mol/L dithiothreitol, and 0.2% bromophenol blue) followed by separation by SDS-PAGE electrophoresis. Proteins were transferred to PVDF membranes and blocked with 5% skim milk powder at room temperature for 1 h. Next, the PVDF membranes were washed with TBST and incubated with primary antibodies against target proteins at 4 ℃ overnight, followed by two washes with TBST. PVDF membranes were incubated with the appropriate secondary antibodies at room temperature for 1 h and washed three times with TBST. The blots were reacted with nitroblue tetrazolium (NBT) and 5-bromo-4-chloro-3′-indolylphosphate (BCIP), or were visualized by using the Azure C500 Infrared Imaging System (Azure Biosystems, Dublin, CA, USA).
-
HEK293T and Hela Cells (20–50% confluent) seeded onto coverslips in 6-well plates were transfected as indicated. 24 h post-transfection, the cells were fixed with 2% formaldehyde and permeabilized with 0.25% Triton X-100, blocked in 10% serum, and incubated with mouse anti-myc mAb (Millipore), mouse anti-flag mAb (Sigma), and mouse anti-V5 mAb (Invitrogen) diluted 1:1000 for 2 h. They were then stained with Alexa Fluor 488-conjugated goat anti-mouse IgG and Alexa Fluor 633-conjugated goat anti-mouse IgG (Molecular Probes) diluted 1:1000 for 1 h. After washing, the coverslips were mounted with mounting medium (Sigma). The images were acquired on a Zeiss LSM710 confocal microscope and adjusted with ZEN software (Zeiss).
-
Data from all cell culture-based assays are presented as the mean ± SD. SPSS 19 was used to evaluate differences among groups. P < 0.05 was considered indicative of a significant difference. ns, not significant (P > 0.05); *, P < 0.05; **, P < 0.01.
Cell Culture and Reagents
Plasmids, siRNA and DNA Transfection
Enzyme-Linked Immunosorbent Assay
HBV Virions Separation from Culture Supernatant
HBV DNA, RNA Extraction and Southern/Northern Blot
Real-Time PCR and Reverse Transcription Real-Time PCR
Luciferase Assay for HBV Promoter Activity
Immunoprecipitation (IP)
Western Blot Analysis
Immunofluorescence
Statistical Analyses
-
Previous studies found that ATP1B3 is involved in the replication of HIV-1 and EV71 (Lu et al. 2016; Nishitsuji et al. 2016). And we recently revealed that ATP1B3 restricted HBV propagation by inducing NF-κB activation (Zheng et al. 2020). However, NF-κB inhibitor could impair the restraint of ATP1B3 on HBV replication by around 50%, suggesting that there may be another mechanism employed by ATP1B3 to inhibit HBV replication. To further confirm the restrictive role of ATP1B3 and to reveal the unknown mechanism of ATP1B3 on HBV suppression, HepG2.2.15 and HepAD38 cell lines stably expressing or inducibly expressing HBV viruses were used. ATP1B3 overexpression significantly reduced the amount of HBsAg by ~ 50% in the supernatants of HepG2.2.15 or HepAD38 cells at 48 h post-transfection (Fig. 1A, 1B, 1G, 1H), and ATP1B3 suppression on HBV is in a time-dependent manner (Fig. 1C, 1I). siRNA-mediated ATP1B3 silencing obtained the opposite effect (Fig. 1D–1F, 1J–1L), further confirming our previous results that ATP1B3 inhibits HBV replication.
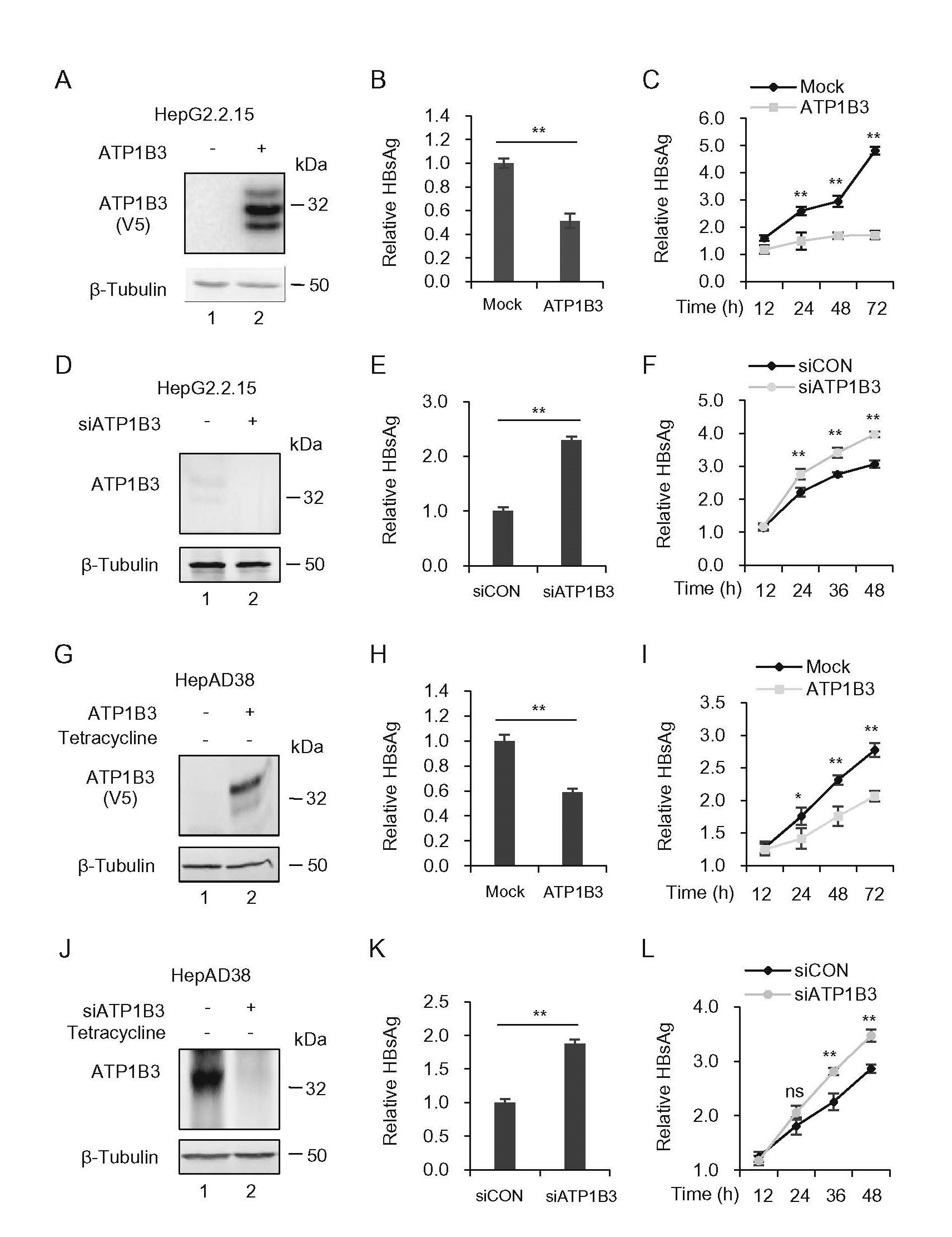
Figure 1. ATP1B3 inhibits HBV replication in HBV-producing HepG2.2.15 and HepAD38 cells. The effect of overexpression (A–C, G–I) or knockdown (D–F, J–L) of ATP1B3 on HBV replication in HepG2.2.15 and HepAD38 Cells. HepG2.2.15 and HepAD38 were transfected with 400 ng ATP1B3 plasmids (A–C, G–I) or siRNA targeting ATP1B3 (D–F, J–L). 48 h post-transfection, ATP1B3 expression was determined by Western blot (A, D, G, J). Relative amounts of HBsAg in supernatants were determined by ELISA (B, E, H, K). HBsAg in culture supernatants was determined by ELISA. Point-in-time analysis of HBsAg in culture supernatants was determined by ELISA (C, F, I, L). The results shown are the means with SD from three independent experiments. ns P > 0.05, *P < 0.05, **P < 0.01.
-
Sequence analysis and homology structure modeling revealed that ATP1B3 contains a small N-terminal intracellular domain (30 amino acid), a TM helix and a larger C-terminal extracellular domain (~ 240 amino acids) (Fig. 2A). In the extracellular domain, there are three conserved disulfide bonds, which are important for forming a stable pump, and two glycosylation sites (Clausen et al. 2017). To determine the functional domains of the ATP1B3 required for HBV restriction, we constructed the N-terminus (∆NT), TM helix (∆TM) and C-terminus (∆CT) truncated mutants (Fig. 2B). HepG2 cells were transfected with the ATP1B3 wild-type (WT) or variants, along with the HBV expression plasmid. The expression of ATP1B3 and mutants were confirmed by Western blot analysis (Fig. 2C) and HBsAg in the cell culture was detected by ELISA. The results showed that the ATP1B3 ∆CT mutant maintained the moderate ability to inhibit HBsAg production (Fig. 2D), although it was defective in NF-κB activation (Fig. 2E), suggesting another unknown mechanism employed by ATP1B3 to inhibit HBV replication.
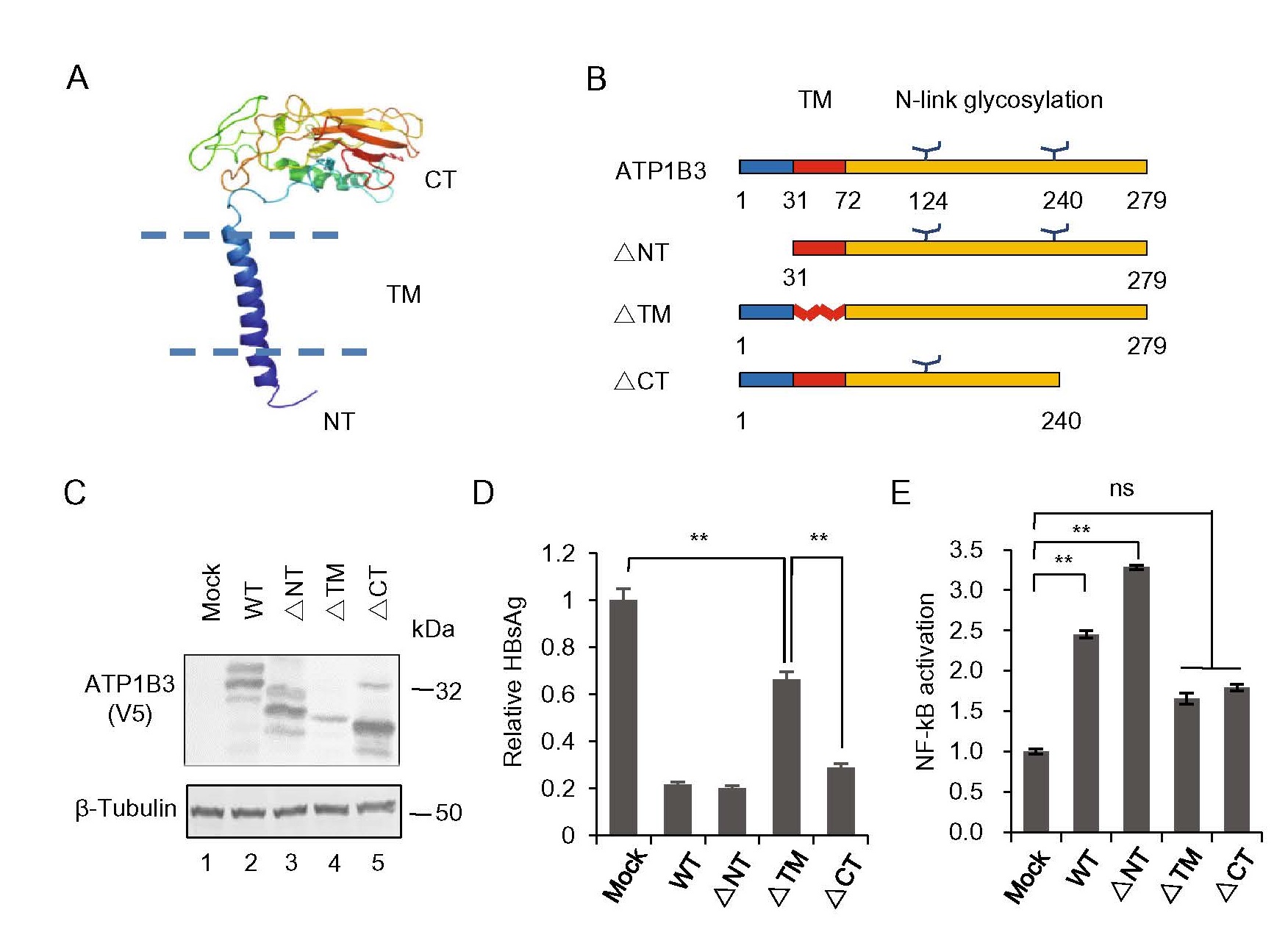
Figure 2. Functional domain of ATP1B3 is required for HBV restriction. A A hypothetic structure model of the wild-type ATP1B3 was generated using SWISS-MODEL. B Three truncated mutations were constructed as indicated. C The wild-type or mutants were transfected and expressed well in HEK293T cells. D ELISA results showed that ∆TM mutant was defective in HBV suppression, but ∆CT maintained the ability to inhibit HBV replication. E Truncated mutations of ∆TM and ∆CT abolished most of the ability to activate NF-κB. **P < 0.01. Error bars are presented as SD.
-
In order to further investigate the above mentioned mechanism, we detected the effect of ATP1B3 on the levels of HBsAg and HBV virion-associated DNA in the supernatant as well as intracellular HBV DNA and RNA levels. The pHBV1.3 and HA-tagged ATP1B3-expressing plasmids were used to transfect Huh7 cells (Fig. 3A). 48 h post-transfection, the levels of HBsAg and HBV virion-associated DNA in the supernatant which were concentrated by IP with polyclonal anti-HBs antibodies conjugated to protein G were determined by ELISA and real-time PCR (Fig. 3B, 3C). The results showed that ATP1B3 expression reduced HBsAg to ~ 45%, and HBV virion-associated DNA to ~ 33% in the culture. However, Southern blot and real time-PCR showed that ATP1B3 had no effect on the levels of intracellular HBV rcDNA, dsDNA, and ssDNA (Fig. 3D, 3E). Northern blot showed that 3.5 kb HBV RNA had not been affected by ATP1B3, but 2.4 and 2.1 kb RNA had been upregulated slightly (Fig. 3F), which is consistent with reverse transcription real time-PCR results in Fig. 3G. The weak activation of ATP1B3 on HBV promoter S1P, S2P, XP suggested that ATP1B3 might activate the transcription of HBV 2.4 and 2.1 kb transcripts (Fig. 3H). Taken together, these data showed that ATP1B3 restrained HBV propagation was not associated with HBV DNA and RNA levels, might be associated with viral proteins levels.

Figure 3. ATP1B3 does not affect the levels of intracellular HBV DNA or RNA. pHBV1.3 plasmids were transfected with empty vector or ATP1B3 plasmids into Huh7 cells for 48 h. A Intracellular exogenous ATP1B3 in Huh7 was determined by Western blot. B HBsAg in culture supernatants of Huh7 was determined by ELISA. C Supernatant HBV virion-associated DNA was determined by real-time PCR. D Intracellular HBV DNA was detected by Sorthern blot. E HBV DNA was measured by real-time PCR. F Intracellular HBV RNA was detected by Northern blot. G Total intracellular HBV RNA was measured by reverse transcription real time-PCR. H Increased doses of ATP1B3 and four HBV promoter fused luciferase reporter plasmids were transfected into Huh7 cells for 48 h, respectively. Intracellular luciferase activity was measured. I HEK293T cells, transfected with pHBV1.3 and empty vector or ATP1B3, were treated with the specific Na+/K+ ATPase inhibitor ouabain as indicated. 48 h post-transfection, HBsAg expression in the supernatant was measured by ELISA. The results shown are the means with SD from three independent experiments. ns P > 0.05, *P < 0.05, **P < 0.01.
In order to investigate whether the restriction of ATP1B3 on HBV is associated with Na+/K+ ATPase activity, the selective Na+/K+ ATPase inhibitor ouabain, a cardiotonic steroid (CTS), was used (Schoner and Scheiner-Bobis 2007). The result showed that the inhibitor ouabain couldn't abolish the restriction of ATP1B3 on HBsAg, although ouabain itself also decreased HBsAg expression to a certain extent, which might due to its broad inhibition activity on Na+/K+ ATPase in cells (Fig. 3I). Therefore, we speculated that ATP1B3 might reduce the amount of supernatant HBsAg in a Na+/K+ ATPase activity-independent manner.
-
We next investigated the levels of intracellular HBV viral proteins in the HepG2.2.15 cells in the presence of ATP1B3 (Fig. 4A). Notably, the intracellular protein levels of LHBs, MHBs and SHBs were reduced by ATP1B3. Since ATP1B3 reduced HBsAg production independent of HBV 2.4 and 2.1 kb RNA transcription, we speculated ATP1B3 might induce the decrease of some HBV viral proteins in the cells. To verify our speculation, plasmids encoding LHBs, MHBs, SHBs, and Core proteins were co-transfected with ATP1B3 into HEK293T cells. Similar to the result in HepG2.2.15 cells, the amount of LHBs and MHBs was obviously reduced in the presence of ATP1B3. SHBs were affected less and core protein basically was not affected by ATP1B3 (Fig. 4B). Protein synthesis inhibitor Cycloheximide (CHX) chase assay confirmed that ATP1B3 expression accelerated the degradation of LHBs and MHBs, whereas SHBs were affected less (Fig. 4C–4F). Proteasome inhibitor (MG132), lysosome inhibitor (Thap) and autophagy inhibitor (Baf-A1) were used to ascertain which pathway is involved in the down-regulation of ATP1B3-mediated HBV envelope proteins. Western blot showed that proteasome inhibitor MG132 obviously suppressed ATP1B3-induced degradation of LHBs and MHBs (Fig. 4G, 4H). Consistent with the results in Fig. 2, ATP1B3-∆CT mutant still had the ability to induce LHBs and MHBs degradation (Fig. 4G), which explained the reason why ∆CT defective in NF-κB activation still has the ability to inhibit HBV.
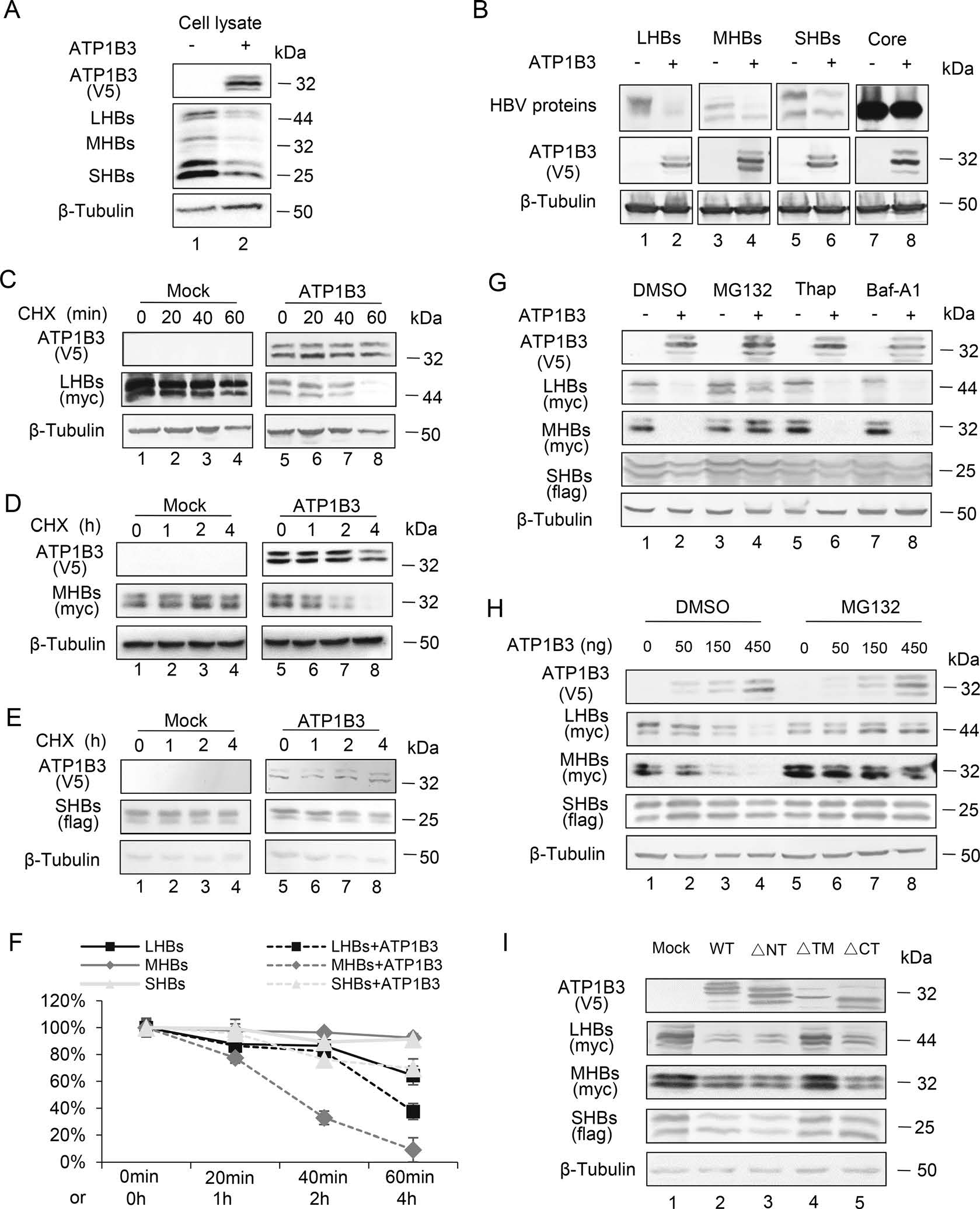
Figure 4. ATP1B3 promotes proteasome-dependent degradation of LHBs, MHBs, and SHBs. A HepG2.2.15 cells were transfected with 300 ng plasmids of ATP1B3. Intracellular LHBs, MHBs, and SHBs were determined by Western blot after 48 h transfection. B 250 ng HBV expression plasmid with empty vector or ATP1B3 plasmid were co-transfected into HEK293T cells. 48 h post-transfection, intracellular expression of HBV proteins and ATP1B3 were determined by Western blot. C LHBs, D MHBs, or E SHBs with empty vector or ATP1B3 plasmid were co-transfected into HEK293T cells. 24 h post-transfection, cells were treated with CHX and point-in-time analysis of LHBs, MHBs, or SHBs was determined by Western blot. F Quantitative analysis of relative LHBs, MHBS, or SHBs level in C–E. G LHBs or MHBs plasmids with empty vector or ATP1B3 plasmid were co-transfected into HEK293T cells. 24 h post-transfection, cells were treated with DMSO, MG132 (10 μmol/L), Thap (3 μmol/L), and Baf-A1(10 nmol/L) for 8 h. Intracellular LHBs, MHBs, or SHBs were determined by Western blot. H Increasing dose of ATP1B3 and LHBs, MHBs, or SHBs plasmids were co-transfected into HEK293T cells. 24 h post-transfection, cells were treated with DMSO or MG132 (10 μmol/L) for 8 h. Intracellular LHBs, MHBs or SHBs were determined by Western blot. I ATP1B3 or indicated mutants were co-transfected with expression plasmids of LHBs, MHBs, or SHBs into HEK293T cells. 36 h post-transfection, cells were harvested and analyzed by Western blot.
-
In order to examine whether ATP1B3 interacts with HBs proteins, IP assay was conducted. The results showed that ATP1B3 specifically interacts with LHBs and MHBs (Fig. 5A, 5B), conversely, co-IP of LHBs and MHBs could pull down ATP1B3 (Fig. 5D). In contrast, the interaction between SHBs and ATP1B3 is weak compared to LHBs and MHBs (Fig. 5C, 5E). On the other hand, ATP1B3-∆TM mutation almost lost the ability to bind LHBs and MHBs. This is consistent with its degradation ability in Fig. 4I.
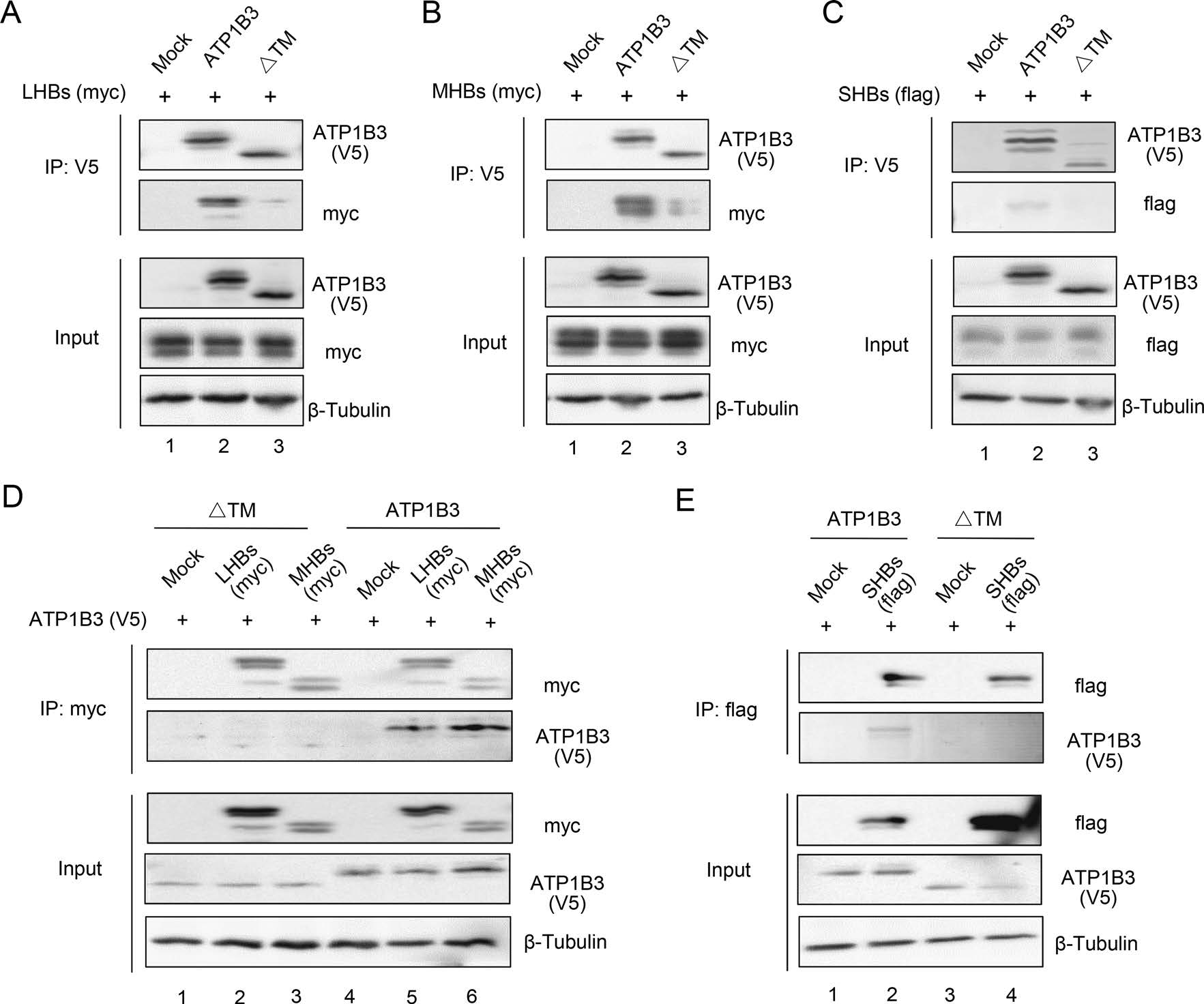
Figure 5. The interaction between ATP1B3 with HBs. ATP1B3 plasmid with empty vector or LHBs (A), MHBs (B), or SHBs (C) plasmids were co-transfected into HEK293T cells. After 48 h culture, IP assay against V5 tag epitope was performed to investigate the interaction between ATP1B3 and LHBs, MHBs, or SHBs. D IP assay against myc tag epitope was performed to investigate the interaction between ATP1B3 and LHBs, MHBs. E IP assay against flag tag epitope was performed to investigate the interaction between ATP1B3 and SHBs. In order to obtain the interaction of ATP1B3 and HBs, 10 μmol/L MG132 were added into cell culture for 8 h to prevent the degradation of HBs at 24 h post-transfection.
Immunofluorescence based co-localization analysis was employed on Hela and HEK293T cells to further investigate the interaction between ATP1B3 and HBs. The results showed that, as a transmembrane protein, ATP1B3 alone localized on the cell membrane. However, ATP1B3 exhibited a punctate distribution around the perinuclear region and also spread in the cytoplasm with LHBs and MHBs when coexpressed with LHBs and MHBs, whereas HBc exhibited an aggregated cytosolic distribution and barely co-localized with ATP1B3. Compared with LHBs and MHBs, the co-localization of SHBs and ATP1B3 was weak. We speculated that ATP1B3 diffused from membrane to cytoplasm due to binding to LHBs and MHBs, which can be degraded forcefully (Fig. 6A, 6C). Co-localization and translocation phenomenon indicated that the interaction between ATP1B3 and HBV envelope proteins was essential for sufficient degradation of HBV proteins whatever spatially and temporally, and also reflected ATP1B3-mediated proteasome-dependent degradation in cytoplasm might not correlate with its Na+/K+ ATPase activity on the cytomembrane. We also investigated the co-localization of HBV proteins and ATP1B3-∆TM mutation. The results showed that transmembrane region deletion almost led to non-localization of ATP1B3 with LHBs, MHBs, and SHBs (Fig. 6B). We speculated that the transmembrane region might be the functional domain responsible for the HBs binding and degradation.
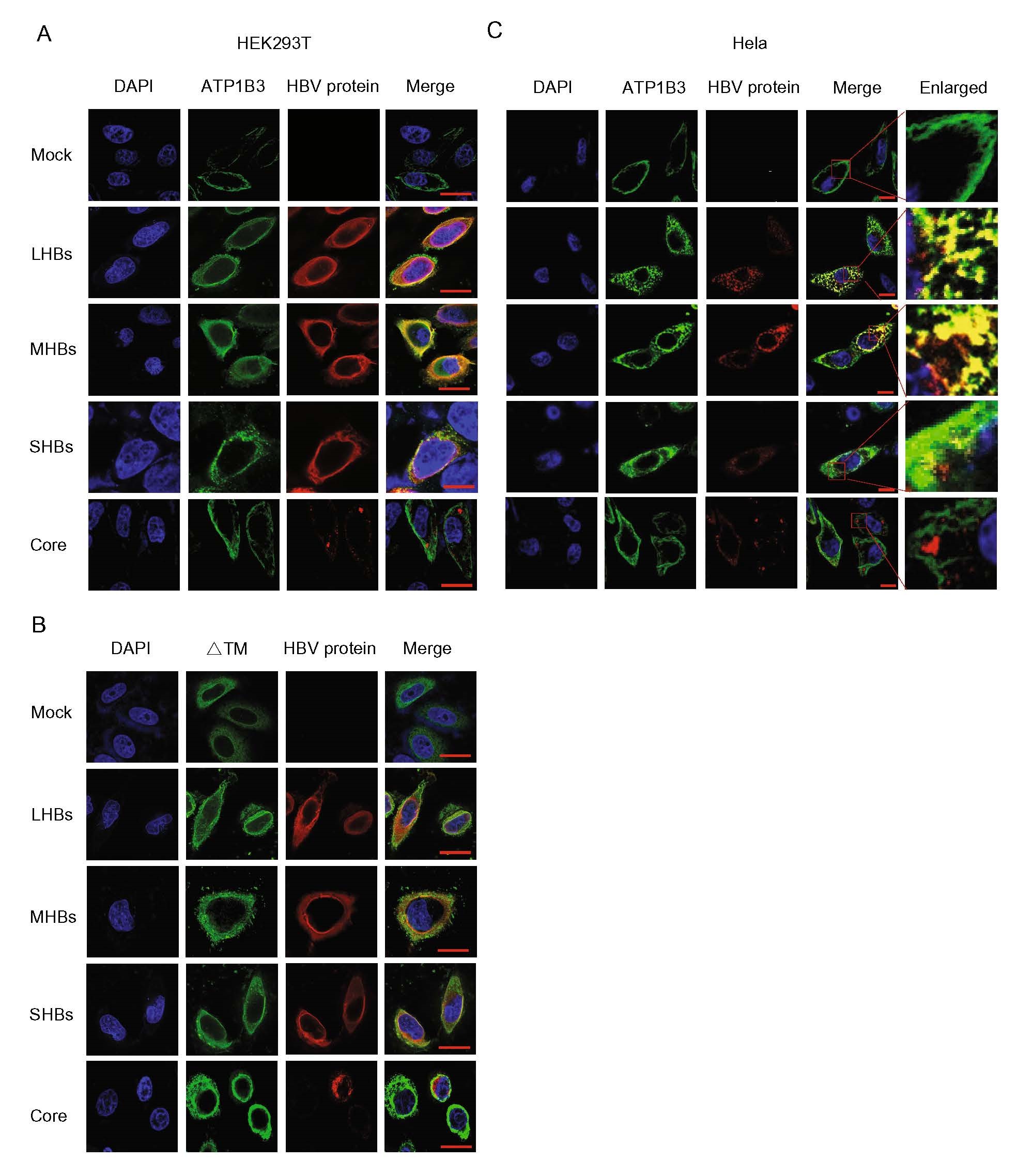
Figure 6. Immunofluorescence based co-localization analysis of ATP1B3 and HBV proteins. A The localization of ATP1B3 with the HBV proteins. ATP1B3 with empty vector or HBV protein (LHBs, MHBs, SHBs, Core) were transfected into HEK293T cells. After 48 h culture, cells were fixed, stained, and imaged. B The co-localization analysis of ATP1B3-∆TM mutation with the HBV proteins in HEK293T cells. C The co-localization analysis of ATP1B3 with the HBV proteins in Hela cells. Scale bars: 5 μm.
-
We next investigated whether ATP1B3 induces LHBs and MHBs polyubiquitination by immunoprecipitating ubiquitin with flag-tagged. The results showed that the degrees of LHBs and MHBs polyubiquitination in the presence of ATP1B3 were stronger than in the absence of ATP1B3 when stained with ubiquitin or targeting proteins antibodies (Fig. 7A, 7B, Lanes 8–9 and 13–14). Interestingly, we observed that ATP1B3 itself was ubiquitinated (Fig. 7A, 7B, lane 16–20). We also investigated which type of ubiquitin linkage was occurring on LHBs and MHBs and found that the ubiquitin mutant that contains only one lysine at position 48 (K48) was sufficient for ATP1B3-mediated HBs ubiquitination, but K63 mutant was not (Fig. 7C, 7D). Moreover, a dominant-negative ubiquitin mutant (Ub-KO flag) which has all seven lysine residues mutated with arginine totally blocked ATP1B3-mediated degradation of LHBs and MHBs (Fig. 8A, 8B), further confirming that ATP1B3-mediated degradation of HBV envelope proteins is a proteasome-dependent manner.
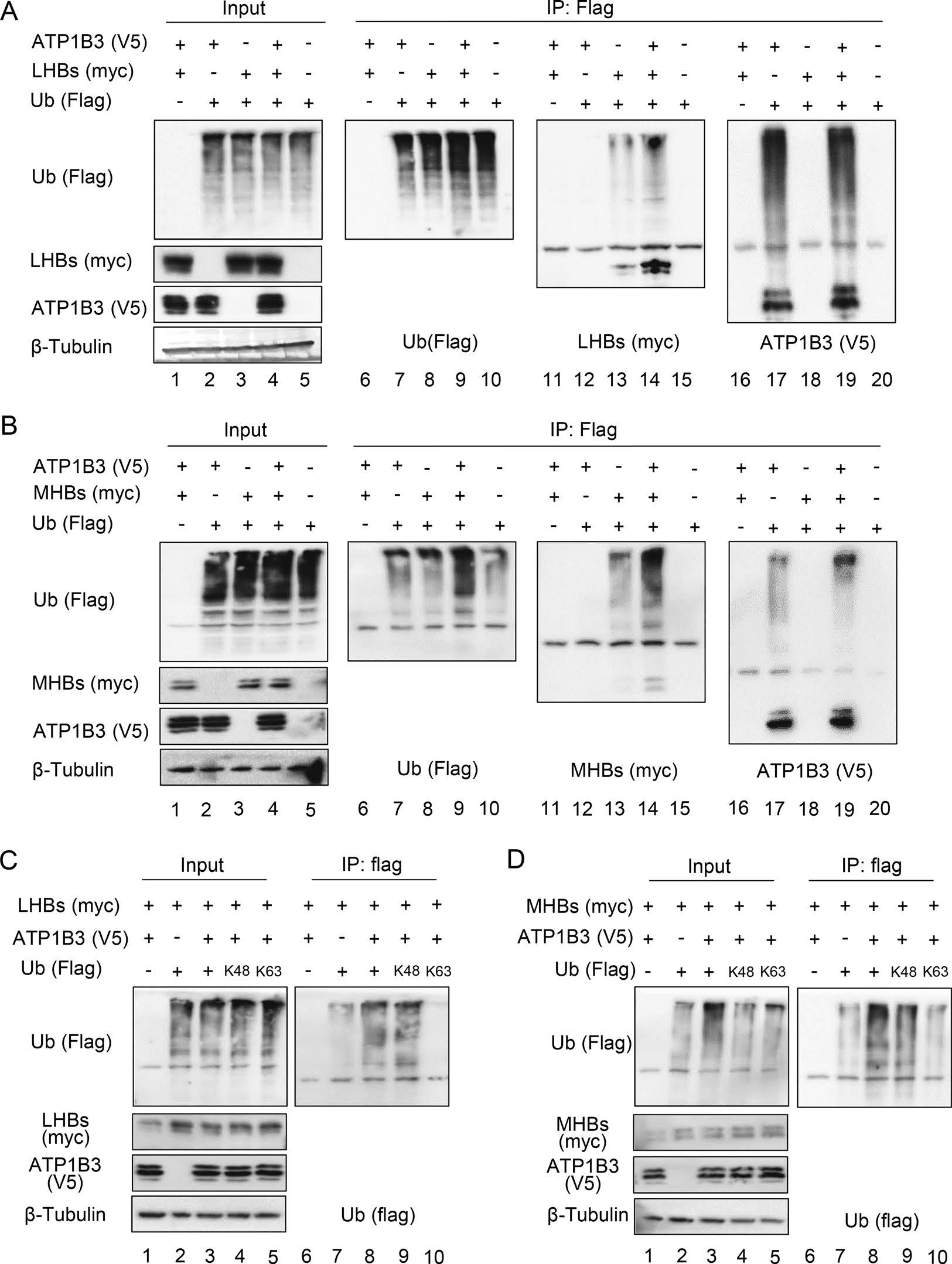
Figure 7. ATP1B3 induces the ubiquitination of LHBs and MHBs via K48-linkage. Immunoblot analysis of LHBs (A) or MHBs-myc (B) ubiquitination in HEK293T cells transfected with constructs encoding various combinations (above lanes) of ATP1B3-V5, Ubiquitin (Ub)-flag, LHBs-myc or MHBs-myc, followed by IP with anti-flag antibody. Immunoprecipitated proteins were probed with flag, myc, V5 antibodies respectively. Immunoblot analysis of ubiquitination of LHBs (C) or MHBs-myc (D) in HEK293T cells transfected with constructs encoding various combinations of ATP1B3-V5, LHBs-myc or MHBs-myc, flag-tagged mutant ubiquitin with substitution of alanine for all lysine residues except the residue noted (K48 and K63), followed by IP with anti-flag antibody. In order to obtain the interaction of ATP1B3 and HBs, 10 μmol/L MG132 were added into cell culture for 8 h to prevent the degradation of HBs at 24 h post-transfection.
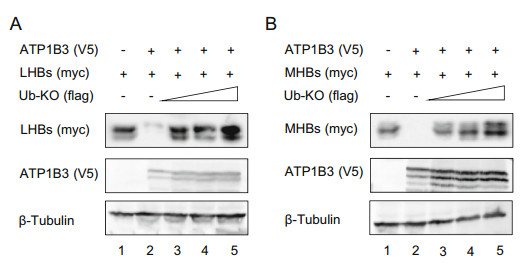
Figure 8. Ubiquitin mutant carrying zero lysines (UB-KO) destroys ATP1B3-mediated degradation of LHBs and MHBs. Immunoblot analysis of LHBs (A) or MHBs-myc (B) in the presence of the increasing dose of Ub-KO. HEK293T cells were transfected with constructs encoding various combinations of ATP1B3-V5, LHBs-myc or MHBs-myc, flag-tagged mutant ubiquitin. 48 h post-transfection, the cells were harvested and probed with indicated antibodies.
ATP1B3 Suppresses HBV Production of HepG2.2.15 and HepAD38 Cells
Identification of ATP1B3 Functional Domains Required for HBV Restriction
ATP1B3 Reduces Supernatant HBsAg and Virion Associated HBV DNA
ATP1B3 Promotes Proteasome-Dependent Degradation of Intracellular HBsAg
ATP1B3 Interacts and Co-localizes with LHBs and MHBs in Cell
ATP1B3 Induces the Polyubiquitination of LHBs and MHBs
-
HBV is a major cause of acute and chronic viral hepatitis. Worldwide, over 450 million people have chronic HBV infection and are at risk for the development of hepatocellular carcinoma (Sagnelli et al. 2020). Due to limited curative treatments for HBV infection, identification of the vital host factors and specific anti-viral mechanisms will help us to develop more precise targeted therapeutic strategies for HBV infected patients.
ATP1B3, the β3-subunit of the Na+/K+ ATPase, not only involves in the Na+, K+ pump activity, but also is part of the functional core of the pump, which is required for trafficking to the plasma membrane (Clausen et al. 2017). Moreover, some pump-independent and evolutionary diverse functions for ATP1B3 have been described. For example, ATP1B3 can regulate T cell activation with no involvement of Na+/K+ ATPase activity (Chruewkamlow et al. 2015). Na+/K+ ATPase also has been reported to be affected by both DNA and RNA viruses, which decrease Na+/K+ ATPase activity or expression (Ulug et al. 1996; Almilaji et al. 2013; Chiu et al. 2014; Chan et al. 2016; Peteranderl et al. 2016). On the other hand, cardiac glycosides which inhibit the Na+/K+ ATPase pump function are effective on various viruses (Dodson et al. 2007; Su et al. 2008; Kapoor et al. 2012), although it is unclear due to changes in ion homeostasis or by activation of intracellular signaling (Xie 2003; Prassas and Diamandis 2008). These findings suggest that ATP1B3 may be involved in broad spectrum antiviral effect. Indeed, ATP1B3 plays an important role in HIV and EV71 replication (Lu et al. 2016; Nishitsuji et al. 2016). Our recent study had reported that ATP1B3 restricts HBV propagation by activating NF-κB/IFN-α (Zheng et al. 2020), but NF-κB inhibitor (Bay11) only partially impaired the restraint of ATP1B3 on HBV replication (~ 50%), hinting that there may be another uncharacterized mechanism for HBV inhibition by ATP1B3. Here, we further demonstrated the inhibition effect of ATP1B3 in two HBV-producing cell models HepG2.2.15 and HepAD38 (Fig. 1) and found ATP1B3 had no effect on HBV intracellular replicative intermediates (HBV DNA) and intracellular transcripts (HBV RNA) (Fig. 3), even there was a slight increase on 2.4 and 2.1 kb HBV RNA production (Fig. 3F). Moreover, ATP1B3 basically had no effect on HBV promoter CP, only caused a slight increase on S1P, S2P, and XP promoters, which are corresponding to a slightly increased transcription of 2.4 and 2.1 kb transcripts (Fig. 3H). ATP1B3 is a type II transmembrane protein, its structure and localization might be closely associated with its anti-HBV function. Thus, we constructed a 3D structural model using ATP1B1 as the template from homology modeling (Clausen et al. 2017), although the three β-subunits (ATP1B1, ATP1B2, and ATP1B3) share low sequence identities (39 ~ 47%). Like ATP1B1, ATP1B3 has a small (~ 30 amino acid) N-terminal intracellular domain, a TM helix, and a larger (~ 240 amino acids) C-terminal extracellular domain. To further evaluate the functional domain of ATP1B3 involved in HBV restriction, three truncation mutants were constructed based on its structure (Fig. 2). Interestingly, the ∆CT mutant showed dramatically decreased activation of NF-κB signaling, but did not fully lose the ability to inhibit HBV. This result indicated that ATP1B3 has an uncharacterized mechanism to inhibit HBV replication.
Ubiquitin proteasome system (UPS) triggered protein ubiquitination is a conserved post-translational modification affecting various biological processes including viral replication. Ubiquitination has multiple effects on viral replication, including viral genome uncoating, viral replication, and immune evasion (Gao and Luo 2006; Luo et al. 2010). Moreover, UPS-mediated viral protein degradation may also constitute a host defense process against viral infections (Luo 2016). Here, we found for the first time that ATP1B3 down-regulates the expression of HBV envelope proteins in a proteasome-dependent manner (Fig. 4), although it is unclear whether ATP1B3 directly induces HBV envelope proteins degradation as an E3 ligase or indirectly down-regulates the expression of HBV envelope proteins by triggering some E3 ligase, which needs to be further investigated in the future. It has been reported that Na+/K+ ATPase is a key scaffolding protein which can interact with signaling proteins such as protein kinase C (PKC) and phosphoinositide 3-kinase (PI3K) (Huang et al. 1997a, b; Mohammadi et al. 2001). If ATP1B3 functions as an E3 ligase, it is possible that ATP1B3 recruits other components to form E3 ligase as a scaffolding protein.
ATP1B3 is broadly expressed on the surface of leukocytes (Chiampanichayakul et al. 2006). As glycoproteins, HBsAg (LHBs, MHBs, and SHBs) all synthesized and folded in the endoplasmic reticulum prior to being exported to the Golgi complex and finally being sent out to the plasma membrane. We infer that ATP1B3 should interact with HBsAg on the endoplasmic reticulum, Golgi, or other membrane structures depending on their transmembrane regions. By IP assay, we confirmed the strong interaction between ATP1B3 and LHBs or MHBs (Fig. 5). Meanwhile, the co-localization analysis showed that part of the ATP1B3 translocated from cytomembrane to the reticular organelles in the cytoplasm in presence of LHBs or MHBs (Fig. 6).
In summary, our work determined that ATP1B3 restricts HBV replication via two mechanisms, one of which is to activate the NF-κB signaling pathway, stimulate IFN-α production. On the other hand, ATP1B3 can also hijack UPS to degrade HBV envelope proteins, which further restricts HBV replication. Our work highlights ATP1B3 as a potential therapeutic molecule in HBV infection. The role of the β-subunit family of Na+/K+ ATPase in virus infection was also suggested to be worthy of further investigations.
-
This work was supported by funding from the National Natural Science Foundation of China (81672004 and 31270202), the Chinese Ministry of Science and Technology (2018ZX10302104-001-010), Science and Technology Department of Jilin Province (20160101044JC and 20190101003JH), Health Commission of Jilin Province (2016J065), the Key Laboratory of Molecular Virology, Jilin Province (20102209), and Norman Bethune Program of Jilin University (2015331) to Baisong Zheng.
-
WZ and BZ contributed to the conception of the study; BZ, JZ, TZ, and XZ performed the experiment; WZ and BZ contributed significantly to analysis and manuscript preparation; WZ and BZ performed the data analyses and wrote the manuscript; HW, ZL, CH helped perform the analysis with constructive discussions. All authors read and approved the final manuscript.
-
The authors declare that they have no conflict of interest.
-
This article does not contain any studies with human or animal subjects performed by any of the authors.

 DownLoad:
DownLoad: