









-
Grass carp reovirus(GCRV) is the first aquatic virus isolated and characterized in China. More than 60 aquareoviruses have been identified since the first reovirus-like virus was isolated from aquatic tissues by Meyers in 1979 (12, 16). Many of these isolates cause asymptomatic infection or infections with very minor diseases. It has been recognized that GCRV is the most pathogenic among all the isolates of aquareoviruses reported to date (16). Therefore, GCRV provides a good model system to study aquareovirus replication and pathogenesis and such studies also have significance in the fish farming agriculture.
GCRV has been classified as a member of genus Aquareovirus of family Reoviridea (13, 19). Similar to other Reoviruses, the virion of GCRV has a multilayered spherical structure enclosing a genome consisting of 11 segments of dsRNA. Due to the lack of standard serotypes, the classification of aqua-reovirus into the Aquareovirus genus is currently done mainly based on RNA:RNA hybridization. According to Rangel et al., six different genetic groups (Aquareovirus genogroups A-F) have been identified among aquareovirus isolates based on reciprocal RNA blot hybridization and those not belonging to genogroups A-F, such as GCRV, are classified into an additional group G (16). More recently, genomic sequence and phylogenic analyses of GCRV and three other aquareovirus isolates have suggested that GCRV should be classified as a member of genogroup C. To date, among the isolated aquareoviruses, GCRV and striped bass reovirus (SBRV, Aquareovirus A) have been characterized in the greatest detail (1, 5, 7, 14, 18). Sequence and phylogenic analyses indicate a relatively higher level of sequence homology between aquareoviruses and mammalian orthoreoviruses compared with other members of the family (1, 5).
While considerable fundamental and applied research has been carried out on mammalian or human isolates from the genus Orthoreovirus and Rotavirus (2, 3, 8, 10, 17, 20), very little progress has been made with members of the genus Aquareovirus. According to the complete genome sequence of GCRV and the result of genome analysis, VP7 encoded by GCRV s10 is the outer capsid protein of the virus. To understand the interaction of GCRV infection with its host cells, it is necessary to express the outer capsid protein of GCRV in vitro. The high level expression of GCRV VP7 protein in Prokaryotic Cells was reported at the first time in this study.
HTML
-
CIK(Ctenopharyngodon idellus kidney)cell was used for proliferating Grass carp reovirus(GCRV). GCRV was isolated and stored in author's laboratory (6, 9).
-
cDNA synthesis kit, enzymes, T7 expression system (pRSET vector) and BL21(DE3) pLysS were purchased from Invitrogen (Invitrogen, Carlsbad, USA). PCR reagent and Taq DNA Polymerase were the product of Perkin Elmer. DNA gel extraction kit and PCR clean-up kit were from V-gene biotechnology limited Company (Hangzhou, China). The PCR primers were synthesized by Saibaisheng Company (Shanghai, China).
-
The prime designation of GCRV s10 was based on GenBank sequence (AF403396). Two PCR primers of GCRV s10 which contain specific restriction enzyme digestion sites depending on the multiple cloning sites supplied by the expression vector pRSET-A. The sense primer : 5'-CAT GGA TCC CCG ATC ATC ACC ACG AT-3', the bases underlined are the enzyme digestion site of BamH Ⅰ; the anti-sense primer : 5'-CGC TGA ATT CGA TGA AAC GAG AGA CC-3', the bases underlined are enzyme digestion site of EcoRⅠ. Thermal cycling parameters were as follows: one cycle of denaturation (94℃, 3 min) followed by 35 cycles of denaturation (94℃, 30 sec), annealing (52℃, 1 min) and extension (72℃, 1 min).The cycling program was ended by an extension step at 72℃ for 15 min. The amplified fragment was purified from agarose gels electrophoresis using the PCR Purification kit.
-
The PCR product was ligated with pRSET-A vector and the ligation product was transformed into CaCl2 treated E. coli DH5α and BL21 (DE3) PLysS competent cells, respectively. The recombinants were identified with both restriction enzyme digestion and PCR amplification as described elsewhere (5). In addition, the identified recombinant plasmid that contained the gene of interest was also sequenced by Invitrogen Biotechnology Inc. (Shanghai, China).
-
The positive recombinant transformant was grown in LB media containing 50μg/mL Amp while shaking 225 r/min at 37℃ and then induced by IPTG. The supernatant and pellet were resuspended in 2 × SDS-PAGE loading buffer after collecting amplified bacteria. 20μL of each of the supernatant and pellet sample were loaded on a SDS-PAGE gel and electrophoresed after boiling for 5 minutes. The samples used for protein component analysis were subjected to 10% Sodium dodecylsulfate polyacrylamide gel electro-phoresis (SDS-PAGE), as described elsewhere (7). Proteins were visualized by using Coomassie brilliant blue R-250 (Sigma, USA).
-
To detect expression of the recombinant fusion protein by western blot analysis, we used both his-tag (mouse) and GCRV antibodies (rabbit) against the appropriate epitope. Briefly, the separated recombinant proteins cell lysate that was subjected to SDS-PAGE (12%) was put on Semi-dry transfer cell following the instrument's instruction, and the alkaline phosphatase coupled rabbit anti-mouse and goat anti-rabbit IgG were used for the second antibody (1:2000). The result was observed by developing with AP substrate solution (NBT/BCIP).
Cell and virus
Reagents
Amplification of interest gene
Cloning and identification of recombinant plasmid
Expression of recombinant protein and SDS-PAGE
Western blotting analysis
-
To obtain the GCRV VP7 gene, a 0.9 kb GCRV s10 segment was amplified by using RT-PCR. The PCR amplified fragment was shown in Fig. 1, which matched well with its predicted value.
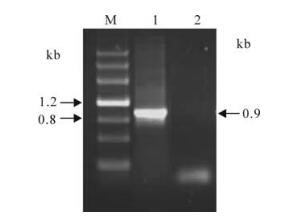
Figure 1. RT-PCR amplification of GCRV s10. M, DNA markⅢ; 1, Positive amplification (GCRV-s10 as template~ 0.9kb); 2, Negative control by using ddH2O as template.
-
To select the required expression product, we chose pRSET-A vector which has the corresponding initiation codon ATG to ligate with the gene of interest. The amplified product and pRSET-A were digested with BamH Ⅰ and EcoR Ⅰ respectively, and then ligated by T4 DNA ligase. The fusion expression vector was named pR/GCRV-VP7. Positive recombinant plasmid pR/GCRV-VP7 was identified by restriction enzyme digestion, PCR amplification and sequencing (data not shown). Fig. 2 shows the electrophoresis image of both PCR and restriction enzyme digestion, which corresponds closely to the predicted GCRV VP7 gene size. Sequence analysis further confirmed the validity of the inserted VP7 gene segments (data not shown).
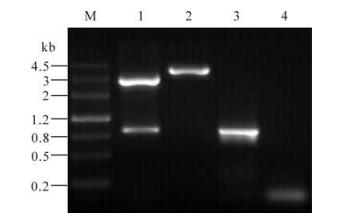
Figure 2. Analysis of recombinant plasmid. M, DNA markⅢ; 1, Double enzymes digested recombinant plasmid with BamHI+ EcoRI; 2, Single enzyme digested recombinant plasmid digested with EcoRI; 3, Positive amplification of PCR (GCRV-s10 as template); 4, Negative amplification of PCR (ddH2O as template).
-
In order to verify the expression of recombinant pR/GCRV-VP7, the recombinant plasmid was transformed into E. coli BL21(DE3) pLysS cells. Fig. 3 shows that the recombinant protein was expressed in E. coli when induced by 1 nmol/L IPTG, and furthermore, Western blot using monoclonal mouse anti-his-tag antiserum showed that expression product can link to anti-his-tag antibody, indicating the recombinant protein is a his-tag fusion protein.
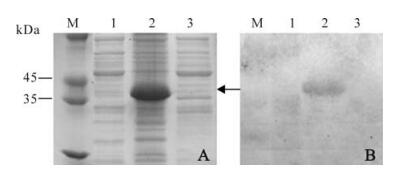
Figure 3. SDS-PAGE analysis of expressed pP/GCRV-VP7 and Western blotting. M, Standard protein marker; A: Coomassie brilliant blue staining. 1, pRSET empty vector as negative control; 2, pR/GCRV-VP7 cell pellets induced by IPTG; 3, pR/GCRV-VP7 cell pellets un-induced; B: Western blot analysis of corresponding to left protein gel samples with anti-his-tag antibody.
-
To obtain the over-expressed recombinant protein, the time course expression of recombinant plasmid pR/GCRV-VP7 was further induced by IPTG at 2, 3, 4, 5 h respectively. SDS-PAGE analysis revealed at the different time points post induction, the VP7 protein was over-expressed with a molecular mass of ~37 kDa (Fig. 4A). Over-expressed VP7 protein accumulated in inclusion bodies and the yield of the GCRV VP7 was about 60% of total bacterial cellular protein content (Fig. 4C). Further western blot analysis showed the expressed fusion protein of VP7 was able to bind immunologically to anti-GCRV serum by comparing with positive control of native GCRV protein (Fig. 4B), indicating that over-expressed fusion product is the recombinant interest protein of GCRV. It may be that the molecular weight of the expressed VP7 protein was a little larger than the native GCRV VP7 protein (~34 kDa), but which is consistent with the expected fusion protein because expression of the interest protein with the N-terminal tag increased the size by approximately 3 kDa.
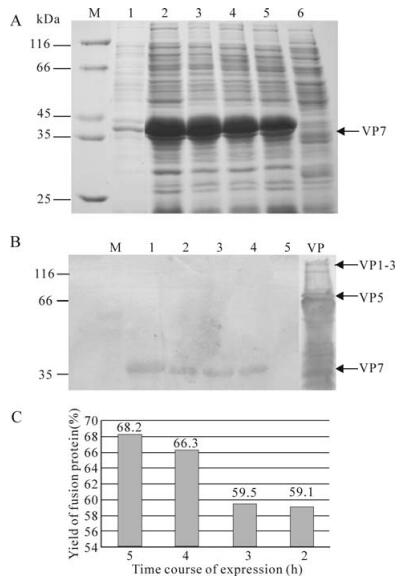
Figure 4. SDS-PAGE analysis of expressed pP/GCRV-VP7 and Western blotting. A: Coomassie brilliant blue R-250 staining. M, Standard protein marker; Lane 1, pRSET-A/GCRV-VP7 supernatant induced 3 h by IPTG; 2-5, pR/GCRV-VP7 pellets induced by IPTG in 5, 4, 3, 2 h respectively; 6, pRSET negative control induced 3 h by IPTG; B: Western blot analysis of corresponding above protein samples with Rabbit anti-GCRV antibody, Lane 1, 2, 3, 4, 5 is matching lane 2-6 in A; VP, Structure protein of GCRV particle; M, pre-stained standard protein marker. C: The yield rate of the expressed fusion protein in the total bacterial cellular protein, which is processed using Glyko Bandscan 4.5software.
RT-PCR amplification of GCRV s10
Construction and identification of the recombinant plasmid
Expression of recombinant VP7 fusion protein in E.coli
Time course expression of the recombinant and western blot analysis
-
The pRSET vectors are pUC derived expression vectors that are designed for high level protein expression and purification from cloned genes in E.coli. High levels of expression of DNA sequences cloned into the pRSET vectors are made possible by the presence of the T7 promoter. The BL21 (DE3) pLysS strain is specifically included for expression of T7 regulated genes. T7 lysozyme is a bifunctional enzyme. In addition to its T7 RNA polymerase binding activity, it also cleaves a specific bond in the peptidoglycan layer of the E. coli cell wall. This activity increases the ease of cell lysis by freeze-thaw cycles prior to purification. For expression of the gene of interest, it is necessary to deliver T7 RNA polymerase to the cells by either inducing expression of the polymerase or infecting the cell with phage expressing the polymerase. In the selected pRSET-A vector, we obtained in frame expression of GCRV VP7. In this case, once sufficient T7 RNA polymerase is produced by adding IPTG, it binds to the T7 promoter and transcribes the gene of interest. In this study we constructed a recombinant GCRV outer capsid protein VP7 gene in pRSET-A vector and successfully over-expressed the VP7 protein in the prokaryotic system.
Sequence analysis and 3D imaging of GCRV revealed that the mature GCRV particle is composed of 7 proteins, VP1-VP7. Among the 7 structural proteins, the VP5 and VP7 proteins comprise the outer capsid shell of the virus. The remaining 5 protein components (VP1, VP2, VP3, VP4 and VP6) are involved in forming the viral core, which is responsible for RNA replication (1, 5). Based on the comparative study between GCRV and MRV sequence, the protein VP5 and VP7 may correspond to u1 and σ3 of MRV. Previous 3D model shows that these two proteins appeared as 200 trimers to form an outer capsid shell. However, VP5 was also the most abundant protein in GCRV, similar to the MRV μ1 protein (7) and it might be located in the intermediate layer of the particle. The VP7 might be the outermost protein and be functional in a protectional role. The trimers on the outer shell might play a role as a receptor recognition site during virus and host cell interaction and entry of virus into host cells (2, 3, 8, 15).
In summary, we have over-expressed GCRV VP7 in Prokaryotic cells. The expressed fusion protein is immunologically related to the epitope of GCRV. A previous study has showed that genomic structure and sequences of GCRV and MRV share a high level of identity, and our result on the expression of recombinant GCRV outer capsid protein VP7 will hopefully provide foundation for future studies aimed at understanding the structure and function relationships during GCRV infection.

 DownLoad:
DownLoad: