











-
The genome of Hepatitis B virus (HBV) only encodes 7 different proteins. The HBV surface and nucleocapsid proteins are structural proteins required for the formation of virions. The HBV polymerase is a multifunctional protein with the terminal protein (TP) domain for priming function, the reverse transcriptase domain for DNA synthesis, and RNAse H domain for removal of pregenomic RNA from the DNA-RNA duplex (10). The HBx protein has diverse regulatory functions and particularly influences the intracellular Ca2+ concentration and therefore activates the Src pathway (1). The role of HBeAg is still elusive. It is proposed that HBeAg may play a role in the induction of immune tolerance. However, the present evidence does not allow to draw a final conclusion. Besides of the functions mentioned above, HBV proteins may directly or indirectly interfere with cellular signaling pathways. The present review will deal with this specific topic with the particular focus on interferon (IFN) signaling. The IFN signaling pathways were of particular interest due to the proposed role of IFNs for the control of HBV infection. Early studies showed that IFN-α is expressed intrahepatically during acute and chronic HBV infection (3, 6, 16, 19). Up to date, IFN-α is the only therapy that could lead to a complete viral elimination (15, 17).
HTML
-
In the 80's, Twu et al found that the transcription of the human beta IFN (IFN-β) gene was suppressed by the 1 828-bp BamHI DNA fragment of HBV (23, 24). By recloning of each complete and partial open reading frame (ORF) present within the 1 828-bp BamHI HBV DNA fragment into a SV40 expression vector, the region encoding HBcAg was identified as the responsible that governs the expression of IFN-β gene (28). Putative proteins encoded by the partial X, P, and S ORFs present in the 1 828-bp BamHI HBV DNA fragment had no effect. A plasmid encoding only the native HBV core antigen (HBcAg), but not one coding for a truncated HBcAg, possessed this inhibitory activity. Furthermore, the inhibition by HBcAg was specific for the regulatory elements of IFN-β gene; none of a variety of viral transcriptional elements was inhibited.
Further, it is reported that HBV downregulates the human IFN-inducible MxA promoter through direct interaction of precore/core proteins (7, 22). The human MxA protein is an interferon (IFN)-inducible GTPase and its antiviral activities has been demonstrated for diverse viruses, particularly negative stranded RNA viruses (14). It is rather controversial whether MxA plays a role in suppression of HBV replication. Gordien et al. could show that the HBV gene expression and replication was reduced in Huh7 cell lines expressing MxA (11). Their data suggested that MxA protein may inhibit the nucleocytoplasmic export of viral mRNA via the PRE sequence. However, IFN-alpha (IFN-α) was able to suppress the HBV replication in MxA-deficient HEp2 cells (a human cervix carcinoma cell line), indicating that MxA is not essential for these activities (21). Thus, the role of MxA in HBV infection is not finally clarified. Nevertheless, MxA expression was reduced in hepatoma cells transiently transfected with the HBV genome (7, 22). Using reporter vectors containing the parts of the MxA promoter, could demonstrate that the IFN-induced expression of the reporter gene (CAT) was inhibited by preC/C expression in a dose-dependent manner. A region comprising IFN-stimulated response elements 2 and 3, upstream of the putative start codon of the MxA promoter appeared to be the target of preC/C proteins and may interact directly with HBV preC/C proteins, as shown by electrophoretic mobility shift assays.
These results demonstrate the ability of HBcAg to interfere with the IFN production and IFN-induced antiviral responses. However, the molecular actions of HBcAg leading to this effect are still not investigated and will be an interesting topic for future research.
-
The N-terminal part of HBV polymerase, known as TP, was found to be essential for the initiation of the replication (25, 26). The TP domain is able to bind to the stem-loop structure of the ε-element on the HBV pregenomic RNA and serves as the primer for the synthesis of the first four nucleotides of the negative strand of the HBV genome. The HBV polymerase and its TP domain, when co-expressed, were able to reduce the reporter gene expression under the control of an ISG (6-16) promoter (8). Cell lines with stable expression of the polymerase were selected on the basis of 2fTGH, a cell line carrying an IFN-inducible marker gene for negative selection. It could be shown that HBV TP domain of the polymerase gene inhibited the response to IFN-alpha. Clones of cells expressing terminal protein alone, selected for the loss of response to IFN-alpha, grew normally and had no detectable response to IFN-alpha, IFN-gamma, or double-stranded RNA. While binding of IFN-alpha to these cells was not impaired, the IFN signaling appeared to be blocked. To verify the relevance of this finding in vivo, the expression of TP in liver biopsy specimens from patients with chronic hepatitis B infection was examined (9). The TP expression was found to be associated with an impaired expression of the interferon-inducible protein beta 2-microglobulin, indicating that the response of hepatocytes to IFN stimulation may be inhibited. Though IFN treatment could significantly reduce the number of cells expressing HBcAg, it did not change the TP expression in hepatocytes.
Recently, an analysis of the gene expression profiles in hepatoma cells with and without HBV infection indicated that HBV blocks the expression of the IFN-inducible myeloid differential primary response protein (MyD88) gene (29). Co-transfection experiments with reporter vectors containing the MyD88 promoter showed that the TP domain of HBV polymerase was responsible for this antagonistic activity. The HBV polymerase protein apparently inhibited the Stat1 nuclear translocation induced by IFN-alpha, but had no effect on the stability and phosphorylation of Stat1. Consistently, HBV polymerase could inhibit the transcriptional activity of other IFN-stimulated response element-driven promoters and the expression of interferon-stimulated genes (ISGs), such as Stat1 and ISG15.
Taken together, HBV polymerase is not only required for viral replication but also for counteraction against the host innate responses.
-
As the HBV gene expression and replication could lead to changes in signaling processes, the cellular gene expression profiles in cells with HBV were investigated with upcoming of the gene array technology. In general, hepatoma cell lines with and without stably integrated HBV genome were compared and the differences in their gene expression profiles were considered (13, 18, 20, 27, 30). Further, cell lines with HBV replication were treated with antivirals like lamivudine and HBV-specific siRNAs (12, 13, 30). The changes of the cellular gene expression profiles under the antiviral treatment were determined.
Since various cell lines and different assays were used, the results of these experiments differed from each other. In addition, the interpretation of the gene array data depends strongly on the specific assay systems and definitions. Therefore, it is no easy to compare the results from different studies or to draw conclusions from the gene array analysis. In general, the presence of HBV gene expression and replication affected only the expression of a limited number of the cellular genes. For example, totally 89 of 14 000 genes included in the gene array were either upor down-regulated ( > 3 fold) in HepG2.2.15 with HBV genomes, taken the gene expression profile in the parental HepG2 as the baseline (27). In addition, there was only a weak functional clustering of differentially regulated genes, indicating that the presence of HBV in stably transfected hepatoma cells does not influence signaling in the upstream parts of specific pathways.
Lamivudine was used to suppress HBV replication in stably transfected cell lines. The suppression of HBV replication by lamivudine shows some influences on the cellular gene expression (12, 13, 30). However, lamivudine is not able to reduce the HBV gene expression in stably transfected cell. Thus, only a small number of genes showed quantitative changes in their expression after the suppression of the HBV. Guo et al. took another approach with HBV specific small interfering RNAs to knock down the HBV gene expression in hepatoma cells (13). The global gene expression changes induced by siRNAs were examined by use of human genome-wide oligonucleotide microarrays. The microarray analysis revealed that the treatment with siRNAs lead the altered expression of cellular genes in HepG2.2.15 cells. Though the patterns of changes of the gene expression differed greatly for the two siRNAs, 18 of these genes were commonly suppressed by both siRNAs. Of particular interest, seven of these genes were expressed at a low level in the parental HepG2 cells. It is assumed that the HBV gene products may up-regulate these genes in HepG2.2.15 cells. As expected, the gene expression profile of HepG2.2.15 cells treated by lamivudine is totally different from that seen with siRNA.
-
Several studies were performed to examine the IFN signaling in the presence of HBV replication. It is clear that HBV does not abolish the stimulation of IFN-stimulated genes (IGSs) in general. HepG2.2.15 cells and other cell lines with the HBV gene expression and replication are able to respond to IFN-α and -γ. Wang et al. found that the induction of ISGs by IFN-α treatment in HepG2.2.15 cells was reduced as compared with HepG2 cells (27). A 24 hour treatment with IFN-α led to changes over 3 fold of 103 genes in HepG2 while 48 genes in HepG2.2.15 showed changes over 3 fold. Some genes like a protein tyrosine kinase were up-regulated in HepG2 but down-regulated in HepG2.2.15. Similarly, Guan et al. found a reduced IFN response in HepG2.2.15 cells, compared with HepG2 cells (12). A treatment with lamivudine could partly restore the responsiveness of HepG2.2.15 cells to IFN stimulation, as demonstrated for two ISGs IFITM1 and 6-16.
Early reports indicated that the expression of the N-terminal part of polymerase (TP) of HBV may impair the cellular responses to interferons (IFN). Therefore, we established HepG2 cell lines stably transfected with a fusion of TP to GFP. The expression of interferon stimulated genes (ISGs) in the cell line was analyzed by gene arrays. The gene array analysis revealed that the expression of 28 cellular ISGs was elevated constitutively without IFN stimulation (Xu et al., unpulished). This finding is similar to the previously published data of Gao et al (13). The transfection of reporter constructs with the promoters of 3 representative ISGs: IFITM-2, -3 and ISG56 confirmed that the presence of TP-GFP acted differently on the gene expression controlled by these promoters. In viral protection assays, these cell lines had a reduced ability to establish antiviral status after stimulation with IFN. Furthermore, the HBV replication occurred efficiently in TP-GFP transfected cells, despite of the up-regulated expression of various ISGs. The enhanced ISG expression in HepG2.2.15 cells and in cells expressing HBV TP is paradox but may provide the explanation for finding using gene arrays. Since the baseline of the ISG expression is higher in HepG2.2.15 cells than in HepG2 cells, the induction of the ISG expression by IFN-α, calculated as fold of the base line expression, is apparently lower in HepG2.2.15.
The mechanisms leading to the changed IFN responsiveness of cell lines with HBV replication is not fully understand. Christen et al. suggested that protein phosphatase 2A (PP2A) may be upregulated in cells with HBV replication (2). PP2A is able to regulate the IFN signaling by inhibiting protein arginine methyltransferase 1 (PRMT1). PRMT1 catalyzes the methylation of stat1 and hypomethylated STAT1 is bound to its inhibitor, PIAS1 and therefore less active. In one hepatoma cell line with HBV replication based Huh7 cells as well as in liver biopsies from patients with chronic hepatitis B, Christen et al. found an increased expression of PP2A and an inhibition of IFN-α signaling. These finding was similar to their previous published data in chronic infection with hepatitis C virus (4, 5). However, this hypothesis needs further confirmation. A different situation was found in HepG2.2.15 for the stat1 function. Guan et al. showed that both the stat1 phosphorylation and the formation of ISGF3 in hepG2.2.15 cells were enhanced (12). Thus, this issue needs be investigated in the future.
-
The available data indicate that the HBV proteins may lead to modulations of cellular IFN signaling as summarized in Fig. 1. We did not deal the function of the HBV x protein in the present review. It is clear that HBV x protein has a broad influence on the cellular processes including different signaling pathways. It will be important to study the functions of single HBV proteins but also to understand their relative role in the context of HBV replication with the presence of HBV proteins. In addition, the findings in cell culture systems need verification for their biological significance in vivo.
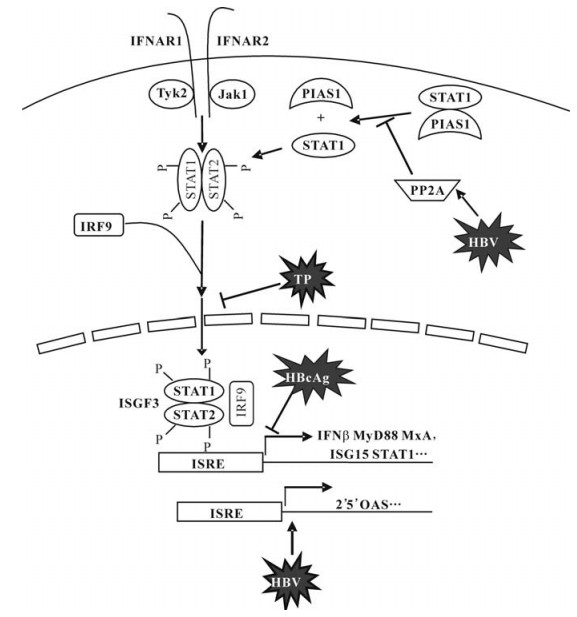
Figure 1. A summary of described interaction of HBV proteins with cellular signaling pathways.

 DownLoad:
DownLoad: