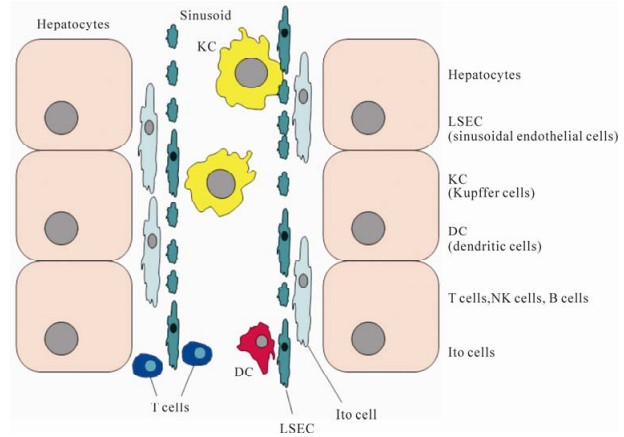
-
Akira S, Takeda K. 2004. Toll-like receptor signalling. Nat Rev Immunol, 4 (7): 499-511.
doi: 10.1038/nri1391
-
Alter H J, Seeff L B. 2000. Recovery, persistence, and sequelae in hepatitis C virus infection: a perspective on long-term outcome. Semin Liver Dis, 20 (1): 17-35.
doi: 10.1055/s-2000-9505
-
Auffermann-Gretzinger S, Keeffe E B, Levy S. 2001. Impaired dendritic cell maturation in patients with chronic, but not resolved, hepatitis C virus infection. Blood, 97 (10): 3171-3176.
doi: 10.1182/blood.V97.10.3171
-
Beasley R P, Hwang L Y, Lin C C, et al. 1981. Hepatocellular carcinoma and hepatitis B virus. A prospective study of 22 707 men in Taiwan. Lancet, 2 (8256): 1129-1133.
-
Bigger C B, Guerra B, Brasky K M, et al. 2004. Intrahepatic gene expression during chronic hepatitis C virus infection in chimpanzees. J Virol, 78 (24): 13779-13792.
doi: 10.1128/JVI.78.24.13779-13792.2004
-
Bröring R, Wu J, Men Z, et al. 2008. Toll-like receptor stimulated non-parenchymal liver cells can regulate hepatitis C virus replication. J Hepatol, in press.
-
Foy E, Li K, Sumpter R Jr, et al. 2005. Control of antiviral defenses through hepatitis C virus disruption of retinoic acid-inducible gene-Ⅰ signaling. Proc Natl Acad Sci USA, 102 (8): 2986-2991.
doi: 10.1073/pnas.0408707102
-
Gorczynski R M, Hozumi N, Wolf S, et al. 1995. Interleukin 12 in combination with anti-interleukin 10 reverses graft prolongation after portal venous immuni-zation. Transplantation, 60 (11): 1337-1341.
doi: 10.1097/00007890-199512000-00024
-
Groux H, O'Garra A, Bigler M, et al. 1997. A CD4+ T-cell subset inhibits antigen-specific T-cell responses and prevents colitis. Nature, 389 (6652): 737-742.
doi: 10.1038/39614
-
Guidotti L G, Ishikawa T, Hobbs M V, et al. 1996. Intracellular inactivation of the hepatitis B virus by cytotoxic T lymphocytes. Immunity, 4 (1): 25-36.
doi: 10.1016/S1074-7613(00)80295-2
-
Guidotti L G, Rochford R, Chung J, et al. 1999. Viral clearance without destruction of infected cells during acute HBV infection. Science, 284 (5415): 825-829.
doi: 10.1126/science.284.5415.825
-
Helbig K J, Lau D T, Semendric L, et al. 2005. Analysis of ISG expression in chronic hepatitis C identifies viperin as a potential antiviral effector. Hepatology, 42 (3): 702-710.
doi: 10.1002/(ISSN)1527-3350
-
Hiasa Y, Horiike N, Akbar S M, et al. 1998. Low stimulatory capacity of lymphoid dendritic cells expressing hepatitis C virus genes. Biochem Biophys Res Commun, 249 (1): 90-95.
doi: 10.1006/bbrc.1998.9089
-
Kato T, Mizokami M, Orito E, et al. 1999. High prevalance of TT virus infection in Japanese patients with liver diseases and in blood donors. J Hepatol, 31 (2): 221-227.
doi: 10.1016/S0168-8278(99)80217-7
-
Kimura K, Kakimi K, Wieland S, et al. 2002. Activated intrahepatic antigen-presenting cells inhibit hepatitis B virus replication in the liver of transgenic mice. J Immunol, 169 (9): 5188-5195.
doi: 10.4049/jimmunol.169.9.5188
-
Knolle P A, Gerken G. 2000Local control of the immune response in the liver. Immunol Rev, 174: 21-34.
-
Li K, Foy E, Ferreon J C, et al. 2005. Immune evasion by hepatitis C virus NS3/4A protease-mediated cleavage of the Toll-like receptor 3 adaptor protein TRIF. Proc Natl Acad Sci U S A, 102 (8): 2992-2997.
doi: 10.1073/pnas.0408824102
-
McCormick C J, Challinor L, Macdonald A, et al. 2004. Introduction of replication-competent hepatitis C virus transcripts using a tetracycline-regulable baculovirus delivery system. J Gen Virol, 85 (Pt 2): 429-439.
-
McHutchison J G, Fried M W. 2003. Current therapy for hepatitis C: pegylated interferon and ribavirin. Clin Liver Dis, 7 (1): 149-161.
doi: 10.1016/S1089-3261(02)00077-6
-
O'Neill L A. 2004. TLRs: Professor Mechnikov, sit on your hat. Trends Immunol, 25 (12): 687-693.
doi: 10.1016/j.it.2004.10.005
-
Racanelli V, Rehermann B. 2006. The liver as an imm-unological organ. Hepatology, 43 (2 Suppl 1): S54-S62.
-
Rehermann B, Nascimbeni M. 2005. Immunology of hepatitis B virus and hepatitis C virus infection. Nat Rev Immunol, 5 (3): 215-229.
doi: 10.1038/nri1573
-
Robek M D, Wieland S F, Chisari F V. 2002. Inhibition of hepatitis B virus replication by interferon requires proteasome activity. J Virol, 76 (7): 3570-3574.
doi: 10.1128/JVI.76.7.3570-3574.2002
-
Sobao Y, Tomiyama H, Sugi K, et al. 2002. The role of hepatitis B virus-specific memory CD8 T cells in the control of viral replication. J Hepatol, 36 (1): 105-115.
doi: 10.1016/S0168-8278(01)00264-1
-
Steinbrink K, Wolfl M, Jonuleit H, et al. 1997. Induction of tolerance by IL-10-treated dendritic cells. J Immunol, 159 (10): 4772-4780.
-
Sumpter R Jr, Loo Y M, Foy E, et al. 2005. Regulating intracellular antiviral defense and permissiveness to hepatitis C virus RNA replication through a cellular RNA helicase, RIG-I. J Virol, 79 (5): 2689-2699.
doi: 10.1128/JVI.79.5.2689-2699.2005
-
Sun Z, Wada T, Maemura K, et al. 2003. Hepatic allograft-derived Kupffer cells regulate T cell response in rats. Liver Transpl, 9 (5): 489-497.
doi: 10.1053/jlts.2003.50091
-
Wesche H, Henzel W J, Shillinglaw W, et al. 1997. MyD88: an adapter that recruits IRAK to the IL-1 receptor complex. Immunity, 7 (6): 837-847.
doi: 10.1016/S1074-7613(00)80402-1
-
Wieland S F, Spangenberg H C, Thimme R, et al. 2004. Expansion and contraction of the hepatitis B virus transcriptional template in infected chimpanzees. Proc Natl Acad Sci U S A, 17; 101 (7): 2129-2134.
-
Wu J, Lu M, Meng Z, et al. 2007. TLR-mediated control of HBV replication by non-parenchymal liver cells in mice. Hepatology, in press.