











-
Rabies virus belongs to the genus Lyssavirus of Rhabdoviridae and infects a variety of mammals including humans and causes fatal viral encephalitis (3). The rabies virus genome consists of single-stranded, unsegmented, negative-sense RNA of about 12 kb, which encodes five viral proteins (3'-N-P-M-G-L-5') and is contained in a bullet-shaped, bilayered envelope(15). The RNA polymerase (L) and phosphoprotein (P) complex with the nucleoprotein (N) form the nucleocapsid (NC), and the matrix protein (M) and the glycoprotein (G) form the inner and outer layers of the envelope, respectively (16).
Currently, through comparing the N, G gene and non-coding region between the G and L genes, known as the G-L intergenic or pseudogene (Ψ) region, the Lyssavirus genus have been resolved into seven genotypes, which can be divided into two major phylogroups with high bootstrap support. Phylogroup Ⅰ comprises Rabies virus (RV; genotype 1), Duvenhage virus (DUVV; genotype 4), European bat lyssavirus 1 (EBLV-1; genotype 5), European bat lyssavirus 2 (EBLV-2; genotype 6), and Australian bat lyssavirus (ABLV; genotype 7). Phylogroup Ⅱ comprises Lagos bat virus (LBV; genotype 2) and Mokola virus (MOKV; genotype 3) (2). Four recently described lyssaviruses (Aravan, Khujand, Irkut and West Caucasian Bat Virus) recovered from bats in Eurasia still await formal classification but may constitute several new genotypes (6).
According to World Health Organization (WHO) estimates, 55 000 deaths due to rabies are reported worldwide every year; the majority of them being in the developing countries of Asia and Africa (12). In China, human rabies cases have been on the rise during the past few years and since 2003 the number of cases each year have been above 2000 with a total of 2 381 cases reported in 2008. Molecular techniques combining amplification by polymerase chain reaction (PCR) and direct sequencing offer a very powerful method for evaluating the epidemiology of viral diseases and therefore could yield a clearer understanding of the origin and transmission patterns of the rabies. Ultimately, data obtained from molecular epidemiological studies can lead to a better understanding and more effective strategies to control the spread of rabies (1).
In this study, we report a full-length genomic sequence of a dog rabies virus (SH06) isolated in China. This street rabies virus genome sequence has been compared with the sequences of all other complete lyssavirus genomes as well as the complete gene sequences of many other vaccine strains available from GenBank (Table 1).

Table 1. Lyssavirus isolates compared in this study
HTML
-
The SH06 sample was isolated from a rabid dog in Shanghai in 2006. This SH06 sample was confirmed as rabies positive by detection of rabies virus antigen using both the direct immunofluorescent antibody (DFA) test (1) and a rapid rabies enzyme immunodia-gnosis (RREID) method employing anti-nucleocapsid and anti-gly-coprotein monoclonal antibodies. Anti-rabies monoclonal antibodies were obtained from Center for Rabies Diagnosis, Wuhan Institute of Biological Products, China (13). A small amount of the tissue was homogenized using PBS and a 10% homogenate was prepared. For isolation of the virus, 100 μL of the homogenate was inoculated into 3-day-old Bab/c mice intracerebrally. After the development of symptoms in the mice the brain was harvested and the presence of rabies viral antigen was confirmed by ELISA and the tissue was stored in the -70℃.
-
Viral RNA was extracted from 0.1 g samples using 1mL TRIzol® (Invitrogen, Carlsbad, CA, USA), following the manufacturer's instructions. In brief, 250μL Chloroform was added into homogenized tissues, mixed gently, and centrifuged at 13 000 r/min for 15 min. The top aqueous layer of about 450μL was collected and RNA was precipitated by using 500μL of isopropyl alcohol and then centrifuged at 13 000r/min for 10 min and the supernatant was discarded. RNA was washed with 1.2 mL of 75% alcohol, centrifuged at 10 000 r/min for 5 min, and the pellet was air-dried. The pelleted RNA was dissolved in 20μL of diethyl pyrocarbonate (DEPC)-treated water, then vortexed softly and frozen at -70℃.
-
Primers employed for the amplification are listed in Table 2. The primers were designed based on the full-length genomes of the PV strain (GenBank accession number NC_001542) and RC-HL (GenBank accession number AB009663) by using freely available primer designing software ("GeneFisher" Interactive PCR primer design software, http://bibiserv.techfak.uni-bielefeld.de/genefisher2/). At the same time, we referenced many known primers (8, 11). The 11 bases in the two UTRs, which have no variation, were used as templates to design the forward and reverse primers to amplify both the 3' and 5' ends, respectively (4, 11).

Table 2. Primers used for amplification and sequencing of the SH06 strain
-
For reverse transcription, 1μL total RNA, 1μL Random Hexamer Primer (0.2μg/μL) and 4.5μL DEPC-treated water were heated at 100℃ for 1 min, and then quickly chilled on ice for at least 2 min. The cDNA was prepared by using 0.5μL Moloney Murine Leukemia Virus Reverse Transcriptase (MMLV-RT) (200U/μL) (TaKaRa), 2μL reaction buffer and 0.5μL dNTP mix (10 mM each) at 30℃ for 30 min, then incubated at 42℃for 90 min, before heat-inactivation of the MMLV-RT Reverse Transcriptase at 95 ℃ for 5 min in DNA Thermal Cycler (Perkin Elmer Cetus).
-
PCR amplification of the defined stretches of the genome was carried out by using different sets of primer pairs. The polymerase chain reaction (PCR) was performed on a DNA Thermal Cycler (Perkin Elmer Cetus) in a 50μL reaction mixture that consisted of 1.5μL cDNA, 5μL 10×Ex Taq buffer, 20pmol of forward and reverse primers, 100μmol/l of each dNTP and 1 unit of Ex Taq (TaKaRa). The initial denaturation was carried out at 94℃for 4 min followed by 39 cycles using initial denaturation at 94℃ for 1 min, annealing at 55℃ for 50 s, and extension at 72℃ for 90 s. The final extension step was performed at 72℃ for 10 min. To confirm the 3' and 5' sequence of the genomic RNA by rapid amplification of cDNA ends (RACE) method we also used 3'-Full RACE Core Set Ver.2.0 and 5-Full RACE Kit from TaKaRa following manufacturer's instructions. PCR products were confirmed visually by GoldView (SBS Genetech) staining of 1% agarose gels after electrophoresis. The PCR products with expected size were purified using a Agarose Gel DNA Fragment Recovery Kit (TaKaRa) following the manufacturer's instructions. The purified product was then ligated directly into the pMD18-T cloning vector system (TaKaRa) at 16℃ for 1 h. The ligated product was transformed into competent E. coli DH5α cells by heat shock method, following the manufacturer's instructions. The transformed colonies were screened by both ampicillin as resistant marker and blue white color selection using X-gal, IPTG containing LB medium.
-
Sequencing of rabies virus genes cloned in the plasmid was carried out with an Applied Biosystems 3770 DNA automated sequencer (Applied Biosystems Inc, Foster City, CA, USA). The complete genome sequences of 22 isolates were used for the multiple alignments and to understand the phylogenetic relationships among isolates (Table 1). The N, P, M, G, L gene sequences and complete genome sequences was modeled for phylogenetic tree reconstructions. Phylogenetic analysis of complete genome sequences, including phylogenetic tree construction and calculation of nucleotide and amino acid identities were performed using MEGA version 4.0 (14). Phylogenetic trees were constructed using the neighbour joining (NJ) method with Kimura evolutionary distance correction statistics (9). The statistical significance of the phy-logenies constructed was estimated by bootstrap analysis with 1000 (NJ) replicates. Bootstrap values above 70% were considered significant (7). Complete nucleotide sequences of the SH06, aG, CTN, PV, HEP-Flury and ERA (reference vaccine strains) were carefully analyzed, which are the current strains used in the production of human anti-rabies vaccines and veterinary vaccine production in China.
Virus isolate
RNA extraction
Primer design
Synthesis of cDNA
Polymerase chain reaction (PCR) and cloning
Sequencing and phylogenetic analysis
-
The complete genome sequences of the SH06 isolate was composed of 11 924 bases (bp). The five structural genes are similar to other rabies viruses, without any insertion and deletion. The open reading frame (ORF) sizes of N, P, M, G and L were 1 353bp (71-1483), 894bp (1515-2408), 609bp (2496-3104), 1 575bp (3316-4890) and 6 387bp (5407-11793) res-pectively. The full genome encodes five structural proteins of N (450 aa), P (297 aa), M (202 aa), G (524 aa) and L (2 128 aa) (Fig. 1). All five genes are initiated with AACA and terminated with poly (A) 7 (Table 3). The non-coding sequence sizes of 3' UTR, N-P, P-M, M-G, G-L and 5' UTR were 70bp (1-70), 91bp (1424-1514), 87bp (2409-2495), 211bp (3105-3315), 516bp (4891-5406) and 131bp (11794-11924), respectively.
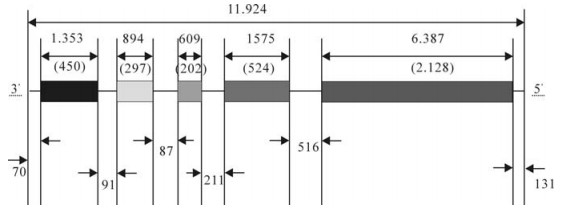
Figure 1. The genome structure of the SH06 strain. Boxes represent open reading frames (ORFs) for five structural proteins, and lines between ORFs show noncoding regions. Numbers in parentheses indicates the number of amino acids in the translated ORF.

Table 3. Transcriptional start and stop signals of the SH06 isolate
-
The comparison of the complete genomic sequence of SH06 with aG, CTN, PV, HEP-Flury and ERA were used for molecular characterization and to understand their relative similarity. The homology of the SH06 with full-length genomes of reference vaccine strains at nucleotide level ranged from 82.2% (with PV) to 86.9% (with CTN). Among the five genes, N gene was the most conserved according to the overall amino acid diversity of the five structural genes, the homology of nucleoprotein gene of SH06 with reference vaccine strains ranged from 84.5% to 88.9% at nucleotide level (nt) and from 94.5 to 98.2% at amino acid level (aa), followed by L gene (nt/aa: 81.9%-87.0%/94.2-97.8%), M gene (85.2%-89.1%/92.8%-97.0%), G gene (78.6%-85.5%/86.8%-92.3%) and P gene (78.6%-86.0% /87.1%-93.0%) (Table 4); the extent of genetic diversity, reflected in percentage identity of five structural proteins, is in the order N > L > M > G > P. Amino acid similarity is dramatically higher than the nucleotide similarity, which means that many of the nucleotide mutations are synonymous.

Table 4. Homology comparisons and overall diversity of nucleotide and amino acid sequences of N, P, M, G, L and complete genome of SH06 with five rabies virus vaccine strains.
-
Phylogenetic analysis was conducted with the NJ method using Maximum Composite Likelihood and bootstrapped with 1000 replicates using software Mega 4.0. To understand the genetic relationships and evolution of rabies virus strains, the entire genome sequence of the SH06 was aligned with 21 complete genome sequences of Lyssavirus Genotype 1 and five structural genes sequences of aG available in the GenBank, Mokola virus was used as the outgroup (Fig. 2). At the same time, the coding regions of N, P, M, G and L gene sequences were aligned using Clustal W, and subjected to tree reconstructions with the NJ method. The topology of the phylogenetic trees generated by the five structural genes was of the same shape, and agreed with phylogenetic tree of the complete genome (not shown).
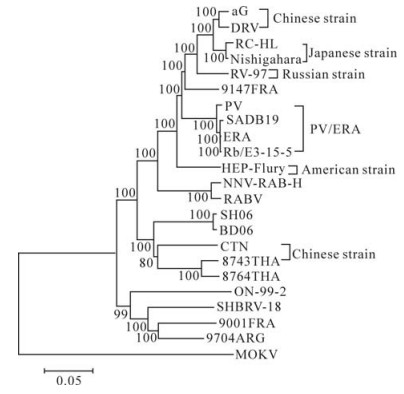
Figure 2. Phylogenetic relationships of 21 complete genome sequences of Lyssavirus Genotype 1 and five structural genes sequences of aG. The Phylogeny was inferred using an NJ method.
Among vaccine strains, six different phylogroups (Fig. 2) could be found, and the grouping was sup-ported by high bootstrap values (BT=100). The Chinese vaccine strains (CTN and aG) forms two different groups. The SH06 is most closely related to Chinese vaccine strain CTN, which is in concordance with previous research (10). At the same time, SH06 is also in the same group as isolates from Thailand.
Genomic structure of the SH06 isolate
Comparative analysis of coding genes sequences
Phylogenetic analysis
-
Though the rabies N and G gene has been used extensively for epidemiological and evolutionary studies, similar analyses were seldom carried out at the whole-genome-scale, especially in China. To gain a better understanding of the molecular characteristics of circulating rabies virus strains in China, we undertook the complete genome sequence analysis of a rabies virus isolate obtained from a rabid dog, and undertook a phylogenetic analysis of 21 complete genome sequences of Lyssavirus Genotype 1 and five structural genes sequences of aG.
The SH06 we studied was closely related to Chinese strains BD06 and vaccine strain CTN, as well as to Thailand strains (8743THA and 8764THA), this was supported by a high bootstrap value (BT=100). However, SH06 was in a different group with other vaccine strains and street strains (Fig. 2). In previous research, the predicted G protein of the CTN vaccine strain and Chinese street rabies virus strains shared a high identity (10). Based on the complete genome sequences analysis alone, the SH06 isolate had 86.9% homology with the CTN vaccine strain but < 82.6% with other vaccine strains at the nucleotide level. Generally, the nucleoprotein and glycoprotein are considered to be the most important for immunogenicity. 97.8% homology of the nucleoprotein and 92.3% homology of the glycoprotein were observed between the SH06 and the CTN vaccine strain respectively. The SH06 shares 86.8%-90% amino acid identity of glycoprotein with other vaccine strains. By analyzing other genes, we observed similar results: SH06 is most closely related to the CTN vaccine strain than any other vaccine strain. Thus, it seems the CTN strain should be most suitable for use in China as a vaccine strain; however, direct vaccine efficiency trials should be undertaken to confirm this hypothesis.
Both the 3'and 5' UTR have conserved signals that play a role to modulate replication and transcription (5). The 3' UTR of the SH06 comprises 70 nt and includes the leader regions potentially transcribed into the leader RNA. Our data also reveals a strict com-plementary sequence of the 11 terminal nucleotides as well as nucleotide positions 13, 14, 15 and 16 from both ends of the genome (Fig. 3). The rabies virus genome consists of a single-stranded RNA virus, but we also found that at least 11 nucleotides of the 3'and 5' UTR sequence of the SH06 isolate are the reverse complement of the sequence; other researches have obtained similar results (3). These observations indicate the genome of the rabies virus is possibly a single-stranded RNA molecule with partial double-strands in the UTR; however, further experiments are necessary to confirm this hypothesis.

Figure 3. Comparison of the 5' and the reverse complementary 3' genomic termini of the antigenomic (+) sense RNA of strain SH06. Identical nucleotides are indicated by a *. TTS: transcription termination signal.

 DownLoad:
DownLoad: