











-
Porcine reproductive and respiratory syndrome (PRRS) is a highly contagious disease causing severe economic losses in the swine industry. It is characterized by reproductive failure, respiratory disease and various clinical signs including loss of appetite, fever, dyspnea and mild neurological symptoms [5, 17]. There are two major PRRSV genotypes [10], referred to as North American and European, and the North American genotype is predominant in China. Previous studies have shown that the non-structural protein 2 (nsp2) coding gene, in particular the central region of this gene, may contribute largely to the genetic variation in PRRSV genomes, while high conservation is exhibited in the N-terminal putative protease domain and the C-terminal predicted transmembrane region [18]. In addition, the Nsp2 protein of PRRSV possesses potential enzymatic function, although this was shown to be highly heterogeneous and variable [15]. Since the first widespread outbreak of PRRS in China in 2006, atypical PRRS, caused by variations in PRRSV strain VR2332, has been reported in more than 10 Chinese provinces and regions [9].
Since the development of loop-mediated isothermal amplification (LAMP) in 2000 by Notomi et al [12], this novel detection method has been extensively used to detect viruses of a wide range of species from humans to animals [4, 14]. This method offers the advantages of simplicity and sensitivity, which can be performed in one step, in real time. Reverse transcription (RT)-LAMP relies on an auto-cycling strand displacement DNA synthesis reaction performed using the Bst DNA polymerase large fragment under isothermal conditions at a temperature of between 60℃ and 65℃, which enables the amplification reaction to be performed in a heating block without the need for precise centralized laboratory facilities [3]. This technique is high specificity because the target sequence is recognized by six independent sequences in the initial stage and by four independent sequences during the later stages of the RT-LAMP reaction. This method generates ascendant turbidity in positive samples, allowing detection by the naked eye or real-time monitoring. Previous reports have indicated that the nsp2 region of the PRRSV genome contains restriction enzyme sites, so in this organism, amplification can be further confirmed by enzyme digestion. Several studies have reported the rapid detection of PRRSV using a RT-LAMP assay [1]. In this study, we developed another RT-LAMP assay to detect prototypic and prevalent North American PRRSV strains that targeted the nsp2 gene. Currently, prevalent PRRSV strains in China are characterized by a 30AA deletion in the nsp2 gene, and can therefore be easily identified by digestion of the amplified product at the characteristic signal peptidase I cleavage site in the nucleotide-depleted gene. Using this RT-LAMP assay, unskilled operators can rapidly and independently detect PRRSV in the field.
HTML
-
Based on the sequences of the prototypic North American PRRSV strain, VR-2332, and 40 other prevalence strains of PRRSV in China published in the GenBank database, six primers (F3, B3, FIP, BIP, LF and LB) were designed for the RT-LAMP assay targeting the two highly conserved terminal portions of the PRRSV nsp2 region. Primer sequences are listed in Table 1.

Table 1. Details of the RT-PCR and RT-LAMP primers designed for the detection of the 5'-untranslated sequences of PRRSV
-
A total of 42 suspected field samples of PRRSV collected from farms in Jiangxi and Guangdong province in 2008. Each specimen was from one piglet or fetus and comprised liver, spleen, lung, lymph node and one or more serum samples. All samples were stored at −80℃ prior to use.
-
Classical swine fever virus (CSFV) strain CHLV, porcine circovirus type 2 (PCV2), Porcine parvo virus (PPV) and porcine rotavirus (PRV), used in the cross-reactivity assay, were kind gifts from Dr Yebing Liu.Prototypic North American PRRSV strain VR-2332, RV68 and SD1-100 isolated and identified by the Control Institute of Veterinary Bioproducts and Pharmaceuticals, China, was used in this study. PRRSV strains GD, BJ, GX, were maintained in our laboratory.
-
Total RNA was extracted from clinical samples or cell culture supernatants of PRRSV isolates using an RNA extraction kit (TaKaRa) according to the manufacturer's instructions.
-
The RT-LAMP reaction was carried out in a total reaction volume of 25 μL containing 12.5 µL of RM and 1 µL of EM from the Loopamp RNA amplification kit (Eiken Co.), 50 pmol of each of the primers FIP and BIP, 5 pmol of each of the outer primers F3 and B3, 25 pmol of each of the loop primers F and B, and 2 μL of the target RNA. The reaction mixture was incubated at 63.5℃ for 60 min in a heating block and was then incubated at 80℃ for 2 min to terminate the reaction.
-
Following incubation at 63.5℃ for 60 min, a 3 μL aliquot of the RT-LAMP amplification reaction was electrophoresed on a 1.5% agarose gel. The gel was then stained with ethidium bromide and visualized on a UV transilluminator at 302 nm. To further differentiate highly pathogenic North American PRRSV strains from the prototypic VR-2332 strain, RT-LAMP amplification products were digested with SupI at 37℃ for 1 h and then analyzed by 1.5% agarose gel electrophoresis. For real-time monitoring of the RT-LAMP assay, the reaction mixture was incubated at 63.5℃ for 60 min in a Loopamp real-time turbidimeter LA-320 (Eiken, Japan). Detection was performed via spectrophotometric analysis by recording the optical density (OD) at 650 nm every 6 s using the Loopamp real-time turbidimeter.PRRSV strains VR-2332, GD, BJ and JX, and water as a negative control, were used as templates and were analyzed by the RT-LAMP assay. Turbidity was assessed, and samples that exhibited an increase in OD of over 0.05 and an OD of greater than 0.25 within 50 min were considered positive. None of the negative controls had a turbidity signal of greater than 0.05 within 50 min.
-
To assess the specificity of the RT-LAMP assay for the detection of PRRSV, DNA of PCV2 and PRV and RNA of PRRSV, swine influenza virus (SIV) and CSFV strain CHLV was extracted and used as the template for the PRRSV RT-LAMP assay. Following DNA and RNA extraction, cross-reactions of PRRSV RT-LAMP with PCV2, PRV and SIV were performed. The reactions were then analyzed with the Loopamp real-time turbidimeter, incubated at 63.5℃ for 60 min.
Isolates GD and VR-2332 were employed to analyze the sensitivity of the RT-LAMP assay for the detection of PRRSV. RNA templates were extracted from 200 μL of MARC-145 cell culture infected with isolates GD or VR-2332. The virus titers of both cultures were 100 times the 50% tissue culture infective dose (TCID50). The RNA template was diluted in a 10-fold series and the reaction mixture was incubated at 63.5℃ for 60 min in a Loopamp real-time turbidimeter.
-
Multiple sequence alignment was conducted using the sequence analysis software Lasergene 1 (DNASTAR Inc., Madison, WI, USA). To distinguish the prototypic North American PRRSV isolate VR-2332 from atypical PRRSVs using the RT-LAMP assay, six primers were designed against the two ends of the variable region of the nsp2 gene of PRRSV. After linearizing by restriction enzyme digestion, the PCR products were gel purified, cloned into the TOPO TA cloning vector (Invitrogen) and sequenced.
-
Total RNA was extracted from the collected field samples and analyzed by the RT-LAMP assay. As a control, a conventional RT-PCR, following the methods of Yue et al. [19], was performed simultaneo usly. To validate the RT-LAMP results, virus was isolated from 42 clinical samples. Porcine alveolar macrophages were used to isolate the virus. Virus isolation followed the methods of Osorio et al [13]. The viruses isolated from samples were tested as outlined by Gonnie Nodelijk et al [11]. Samples were considered negative after two blind passages.
Primers
Clinical samples
Viruses
RNA extraction
RT-LAMP assay
Detection of RT-LAMP amplification
Sensitivity and Specificity of the RT-LAMP assay for the detection of PRRSV
DNA sequencing
Evaluation of the RT-LAMP assay
-
The RT-LAMP assay was standardized using PRRSV strain BJ, isolated from a patient with clinically diagnosed PRRS. The RT-LAMP assay successfully amplified the target sequence of the PRRSV nsp2 region and this was confirmed by agarose gel electrophoresis (Fig. 1). The amplification products appeared in a ladder-like pattern on the gel due to the formation of a mixture of stem-loop DNAs of various stem lengths and cauliflower-like structures with multiple loops formed by annealing between alternately inverted repeats in the target sequence in the same strand. In the negative control, no specific amplification products were observed, in accordance with the results collected by the Loopamp real-time turbidimeter (Fig. 2).
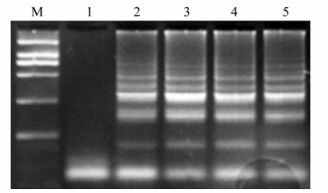
Figure 1. Agarose gel electrophoresis of RT-LAMP products. M, DL2000 Plus Marker; 1, Negative control; 2, PRRSV strain VR-2332; 3, PRRSV strain GD1; 4, PRRSV strain JX1; 5, PRRSV strain BJ1.
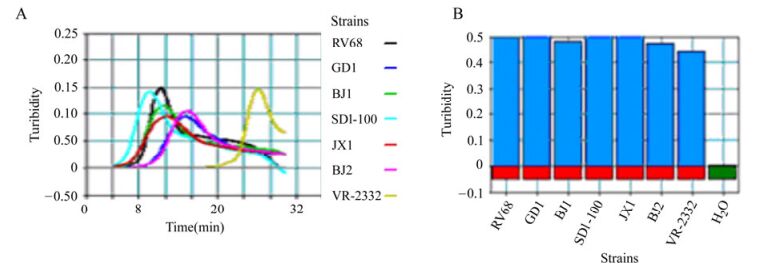
Figure 2. RT-LAMP analysis using a real-time turbidimeter. A: The real-time monitoring results.
-
The RT-LAMP products were digested with SupI and analyzed by 1.5% agarose gel electrophoresis. For strain VR-2332, the RT-LAMP product was digested into two bands of 160 bp and 120 bp, whereas the RT-LAMP product of the highly pathogenic isolate BJ was not digested by SupI (Fig. 3).

Figure 3. Restriction analysis of the RT-LAMP products. M, DL2000 Plus marker; 1, PRRSV strain GD; 2, Enzyme digestion of the RT-LAMP product of strain GD.
-
The sensitivity of the RT-LAMP assay for the detection of PRRSV North American strains was determined using 10-fold serial dilutions of virus that had previously been quantified by the TCID50 assay and comparing these results with those of conventional RT-PCR. The RT-LAMP assay was able to amplify the virus in 60 min, with a detection limit corresponding to a 10–5 dilution of template RNA (Fig. 4). On comparing the two assays, RT-LAMP was found to be 100-fold more sensitive than RT-PCR, which had a detection limit of 0.1 TCID50 of virus (data not shown). The specificity of the RT-LAMP primers for the nsp2 region of PRRSV was determined by checking the cross-reactivity of the primers with other serologically-related viruses, including CSFV strain CHLV and PCV2. The PRRSV-specific RT-LAMP primers demonstrated a high degree of specificity for the North American strains of PRRSV by amplifying the two ends of the PRRSV nsp2 region and yielding negative results with all other viruses tested. The identity of the amplified products was verified by sequencing.
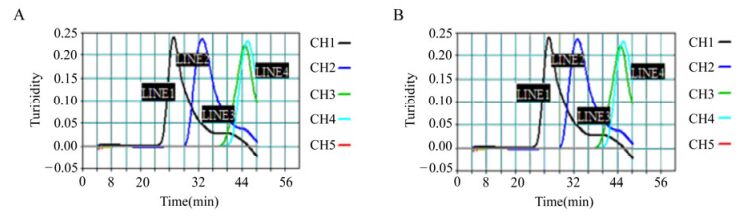
Figure 4. Sensitivity of the RT-LAMP assay for the detection of PRRSV. Serially diluted North American strains of PRRSV, which had previously been quantified by the TCID50 assay, were detected using the LA-320 turbidimeter. A: Strain VR-2332, CH1-CH5 corresponds to dilutions 10–1–10–5. B: strain GD, the dilutions were the same as for A.
-
Following digestion with SpuI, DNA fragments were cloned and sequenced. Sequence analysis indicated over 97% homology between the sequence of the amplified fragment and the nucleotide sequence published in the NCBI database, indicating that the RT-LAMP assay was highly specific.
-
The applicability of the RT-LAMP assay for the clinical diagnosis of PRRSV was validated using acute-phase PRRS-suspected samples collected from highly epidemic areas of China, such as Jiangxi and Guangdong in 2008, and the results were compared with those of conventional RT-PCR. A total of 42 clinical samples were used in this study. The comparative evaluation of these two methods revealed a good correlation between the RT-LAMP assay and conventional RT-PCR at detecting viral RNA. RT-LAMP detected 33 positive samples and RT-PCR detected 30 positive samples among the 42 samples; therefore, the sensitivity of RT-LAMP relative to RT-PCR was 91%. The specificity of RT-LAMP relative to RT-PCR was 100%.
Virus isolation (confirmed by an immunoperoxidase monolayer assay) was performed with all 42 field samples. Virus isolation detected 33 positive samples, the same number of positive samples detected by the RT-LAMP assay. Therefore, these two methods showed 100% sensitivity and 100% specificity (Table 2).

Table 2. Detection of PRRSV in 42 clinical samples based on RT-LAMP, RT-PCR and virus isolation.
Identification of PRRSV isolates using the RT-LAMP assay
Restriction digestion of the RT-LAMP products
Sensitivity and specificity of the RT-LAMP assay
Sequence analysis
Evaluation of the PRRSV RT-LAMP assay with clinical samples
-
There are three methods commonly used for virus detection: virus isolation, serological testing and the detection of genomic sequences by nucleic acid amplification. Virus isolation is the conventional diagnostic method for PRRSV infection; however, these viruses cannot always be successfully isolated from clinical specimens often because specific-pathogen-free porcine alveolar macrophages, the permissive cells for PRRSV propagation, are not available and other cell lines are generally less susceptible to the virus [16]. Serological tests, such as enzyme-linked immunosorbent assays (ELISAs) and immunofluorescence assays, generally require complicated procedures and expensive antibodies and are therefore not widely applicable, especially for early or pre-clinical diagnoses [2, 7, 8]. Although reverse transcription polymerase chain reaction (RT-PCR) is a highly sensitive and specific method, the dependence on specialized equipment limits its use in the field or in basic laboratories [2, 6]. The development of molecular detection methods to simplify the diagnosis procedure is of great importance. RT-LAMP is a one-step assay that does not require a cDNA synthesis step, simplifying the method and decreasing the risk of contamination [18]. The use of three primer pairs corresponding to six target sequences in the RT-LAMP assay was conferred high specificity but can also make primer design a challenge in viruses harboring many mutations in their genome. In the PRRSV RT-LAMP assay, positive amplification resulted in a ladder-like pattern after agarose gel electrophoresis, unlike conventional PCR in which a specific fragment was amplified. To verify positive RT-LAMP results, enzyme digestion, sequencing and real-time monitoring were conducted. Restriction analysis enabled differentiation of the prototypic VR-2332 strain of PRRSV from highly pathogenic isolates of PRRSV, and also digested complex amplification products into stable nucleic acid fragments, facilitating sequencing of this DNA. The high efficiency of the RT-LAMP assay meant that an amplification product could be produced even when the reaction rate was low, resulting in false positives. By employing a Loopamp real-time turbidimeter, the sensitivity and specificity were quantified, resulting in the detection efficiency to be increased and the procedure to be simplified. The sensitivity of the RT-LAMP assay for the detection of PRRSV strains VR-2332 and GD showed that decrease in the concentration of RNA template affected the start point of the reaction, but not the cutoff value of the reaction rate. The curves obtained differed between viral serotypes and genotypes, indicating that virus strains could be discriminated according to their curve characteristics.
The PRRSV RT-LAMP assay described in this study showed greater specificity than conventional RT-PCR. There was no cross-reactivity between PRRSV and other related viruses, such as CSFV strain CHLV, PCV2, PRV and SIV, which suggeated that the method was highly specific. In addition, RT-LAMP is a simple and rapid procedure, providing results within 1 h, which is significantly quicker than conventional RT-PCR. Of the 33 positive samples detected by RT-LAMP, three were missed by RT-PCR. Furthermore, restriction analysis allowed for the specific detection of highly pathogenic PRRSV strains. In summary, we have constructed a rapid, sensitive RT-LAMP assay for PRRSV detection that has the potential to be applied to on-the-spot detection in the field.

 DownLoad:
DownLoad: