











-
The ovine genome contains at least 20 copies of endogenous beta retroviruses (enJSRVs) that are highly homologous with Jaagsiekte sheep retrovirus (exJSRV), an exogenous retrovirus which is responsible for ovine pulmonary adenocarcinoma (OPA)[1, 7, 8, 13, 27]. This infectious tumor naturally involves two types of secretary epithelial cells of the lung, alveolar typeⅡcells and non-ciliated bronchiolar epithelial cells[15, 17]. The cellular receptor for both exJSRV and the enJSRV is hyaluronidase 2 (Hyal-2) and JSRV interacts with target cells by competing the membrane receptor with enJSRV[19, 25]. The relationship between exJSRV and enJSRV is still unclear. It is assumed that endogenous retroviruses (ERVs) are derived from integration events during evolution of ancient exogenous retroviruses into the germline of host animal species. ERVs are transmitted through the germline as stable Mendelian genes, and their structure and sequence are similar to infectious exogenous retroviruses. Some study reports revealed that JSRV genomes share 90–98% homology with enJSRV in deduced amino-acid sequence[1, 2, 17, 21]. The high level of homology makes it impossible to detect any circulating antibody in peripheral blood[12, 24]. One possible explanation for this phenomena is that the existence of enJSRV during the embryonic development periods makes the host tolerant towards exJSRV infection. However, the tolerance mechanisms of the disease are poorly understood, and the research on OPA moves forward slowly.
OPA represents a good model for studying the interactions between endogenous retroviruses and their exogenous counterparts. It is also valuable for research into human bronchioloalveolar cancer (BAC) since many clinical, radiological and histopathological features are similar in both diseases[10].
In order to analyze the establishment of immuno logical tolerance during the embryo period, we evaluated the expression of the enJSRVs envelope (env) protein and Hyal-2 mRNA in the immune organs of fetuses and lambs by In Situ Hybridization and Real-Time reverse transcription PCR methods. The data obtained in the present study provide useful information for the understanding of the immune tolerance pathogenesis.
HTML
-
For the experiments, the Animal Center of Inner Mongolia Agricultural University offered 18 Mongolia ewes which were bred under the same nutritional condition, were oestrus at a scheduled time and became pregnant. Fetuses on day 70 of gestation, fetuses on day 130 of gestation and 3 day old newborn lambs were assigned randomly to be necropsied (n=6 ovines/period). The lungs, thymus, spleen and mesenteric lymph nodes were collected at postmortem examination, snap frozen in liquid nitrogen and stored at -80 ℃ for RNA extraction. Total RNA was isolated from tissues according to the manufacturer's recommendations (RNA extraction kit, TaKaRa, Dalian, China). The integrity of RNA was examined by gel electrophoresis in a denaturing 1.0% agarose gel and the RNA was quantified at 260 nm in a micro-spectrophotometer at an OD 260/280 ratio of 1.8 to 2.0 for all samples, and then stored at -80 ℃. All tissues were also fixed in fresh 4% paraformaldehyde for 10 h, dehydrated through a graded series of alcohol to xylene, embedded in paraffin and cut at 5 μm for In Situ Hybridization.
-
Primers and probes used were designed on the basis of published enJSRV, Hyal-2, β-actins′ sequences and synthesized by TaKaRa Biotechnology Co., Ltd. (TaKaRa, Dalian, China). The information of objective genes is shown in Table 1.

Table 1. Primers and probes used in this study
-
Synthesis of DNA was carried out in 50 μL reaction volumes as follows. 0.5 μL PrimeScript RTase(for 1 Step), 1 μL RNase Inhibitor (40 U/μL), 1 μL TaKaRa Ex Taq HS(5 U/μL), 2 μL dNTP Mixture(10 mmol/L each), 5 μL 10×One Step RT-PCR Buffer, 1 μL One Step Enhancer Solution, 1 μL Forward Primer(20 mmol/L), 1 μL Reverse Primer(20 mmol/L), Total RNA, and RNase free dH2O. The Conditions were 50 ℃ for 30 min, 94 ℃ for 2 min, and 45 cycles of 94 ℃ for 30 s, 30 s annealing (enJSRV 55 ℃, Hyal-2 60 ℃, and β-actin 58 ℃) and with a final extension of 72 ℃ for 1 min. RNase free dH2O in RT-PCR kits were used instead of template RNA as control. The PCR products were purified by Gel Extraction Kit (TaKaRa, Dalian, China) according to the manufacturers' instructions.
A portion of the purified PCR products was separated in a 1.5% agarose gel and visualized by ethidium bromide staining using an imaging system. The density and A260/A280 were analyzed with a micro ultraviolet spectrophotometer to identify the relative expression quantity of the target gene. Total RNA of all samples were used in RT-PCR experiments. The RT-PCR products were sequenced and analyzed with NCBI Blast. At the same time, two sense probes were synthesized according to the sequences of enJSRV and Hyal-2. Then the RT-PCR products of thymus of 70 d fetuses were used to prepare anti sense probes.
The procedure was performed as follows: 1 μg purified template DNA and RNase free dH2O were added to a final volume of 16 μL to a reaction vial, the DNA was denatured by heating it in a boiling water bath for 10 min and then quickly cooled in an ice/water bath. 4 μL DIG-High Prime was added to the denatured DNA, then mixed by centrifuging briefly. The mixture was incubated for 20 h at 37 ℃, then 2 μL 0.2 mol/L EDTA (pH 8.0) was added to stop the reaction. 1 μL labeled probes and labeled control DNA were applied to the nylon membrane. The nucleic acid was fixed to the membrane by baking for 30 min at 120 ℃. The membrane was then transferred into a plastic container with 20 mL maleic acid buffer, and incubated by shaking for 2 min at 20 ℃, incubated for 30 min in 10 mL blocking solution, incubated for 30 min in 10 mL antibody solution, washed with 10 mL washing buffer for 2×15 min, equilibrated 5 min in 10 mL detection buffer, incubated in 2 mL freshly prepared color substrate solution in a appropriate container in the dark. The reaction was stopped by washing the membrane for 5 min with 50 mL RNase free dH2O. Results were documented by digital photography.
-
The procedure was performed as follows: The slide (5 μm) was dewaxed through xylene, dehydrated and digested with protease K/0.1 mol/L PBS (pH 7.4) for 1-2 min. Glycine was added to inhibit digestion, the slide was washed twice with PBS and 4% paraformaldehyde/0.1 mol/L PBS respectively. 20 μL pre-hybridization mixture (5×SSC, 5×Denhardt, 50% Deionized formamide, 1% SDS, 200 μg/mL Salmon sperm DNA) was added and pre-hybrided for 1 h at 42 ℃. The pre-hybridization mixture was removed and replaced with hybridization mixture (5×SSC, 5×Denhardt, 50% Deionized formamide, 1% SDS, 250 μg/mL Salmon sperm DNA, 10% Dextran sulfate, probe)for 18 h at 42 ℃. The sample was then washed as follows: SSC, closed fluid, anti-digoxigenin AP fluid, malefic acid buffer solution, colored buffer solution, NBT/BCIP colored solution, RNase free dH2O, and counterstained with eosin, dehydrated through a graded series of alcohol to xylene and protected with a cover slip. In the negative control, the sections were analyzed by In situ hybridization with DIG-labeled sense probes.
-
First strand cDNA synthesis was performed according to the protocol suggested for the Reverse Transcription System (TaKaRa, Dalian, China) using Oligo(dT)16 primers. Quantification of mRNA levels of the genes was achieved using a Premix Ex TaqTMkit (TaKaRa, Dalian, China) using β-actin as the housekeeping gene. PCR amplification was run using IQTM5 Multicolor Real-Time PCR Detection System (BIO-RAD, America) and carried out in 25 μL reaction volume containing 2 μL cDNA, 12.5 μL SYBR Premix Ex Taq (TaKaRa, Dalian, China), 0.5 μL Forward primer (2 μm), 0.5 μL Reverse primer(2 μm), 2 μL TaqMan probes (0.2 μm) and 8.5 μL RNase free dH2O to make a total volume of 25 μL. The primers and probes used were synthesized by TaKaRa Biotechnology Co., Ltd. (TaKaRa, Dalian, China) and are shown in Table.1. All PCR reactions were run in duplicate and were performed with 45 cycles, including a negative control consisting of PCR-grade water. Data analysis was performed using a relative standard curve and based on the ratio of the fluorescent change observed with the target gene to the fluorescent change observed with β-actin. The expression level was analyzed by one-way ANOVA followed by Duncan's test. The values were expressed as mean ± SD (n = 6).
Preparation of samples
Primer Design and Synthesis
RT-PCR and Preparation of Probes
In Situ Hybridization
Real-Time Reverse Transcription PCR and Data Analysis
-
The values of A260/A280 were in 1.8-2.1, indicating that the total RNA can be used in the analysis of mRNA. Total RNA of all samples were used in RT-PCR experiments, RT-PCR products were separated in a 1.5% agarose gel and visualized using ethidium bromide (Fig. 1). The products of enJSRV and Hyal-2 were sequenced, and the sequences obtained were compared with other published sequences in NCBI GenBank. The sequences were more than 95.9% identical to other published enJSRV sequences (AF136224; EF680297; EF680300; EF680313; EF680317) and more than 98% identical to other published Hyal-2 sequences (NM-174347; NM-001009754; BC102042; BT021810; BT026155). The Results indicated that enJSRV and Hyal-2 mRNAs can be detected in thymus, spleen, mesenteric lymph nodes and lungs of different periods.
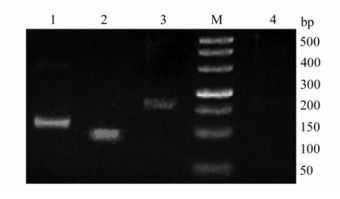
Figure 1. Results of RT-PCR. The 500 bp marker (M) is shown on the far right. lane 1, Hyal-2, lane 2, enJSRV, lane 3, β-actin, lane 4, negative control. Identified bands are approximately 111 bp, 92 bp, 159 bp for lanes 1, 2 & 3 respectively. The RNA used for the RT-PCR experiments was from the thymus of 70 d fetus.
-
In Situ Hybridization revealed clear positive signals in all tested tissues (Fig. 2, Fig. 3, Fig. 4). The mesenteric lymph nodes of 70d fetus were not obtained. With regard to the thymus, positive reactions were observed in thymus lobules, especially in the cortico-medullary junction and medulla of lobules. Similar results were observed in mesenteric lymph nodes and the expression of enJSRV and Hyal-2 mRNA mostly appears in the lymphocytes. The spleen revealed signals which were mainly located in white pulp which contains confertim lymphatic tissue. Signals above background were also observed in the bronchial epithelial cells and alveolar cells of the lungs. In the negative controls, all tested tissues did not show any positive signals when the sections were analyzed with DIG -labeled sense probes. In the negative tissue controls (some tissues of mouse) we also did not detect positive signals (Fig. 5), thus confirming the specificity of the tests.
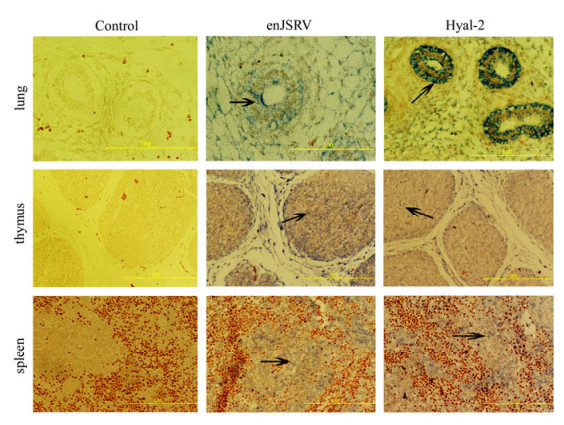
Figure 2. The expression of enJSRV and Hyal-2 in the tissues of 70 d fetuses by In situ hybridization. Cross sections of the lung, thymus, and spleen from ovine fetus hybridized with DIG-labeled probes. The lung bronchial epithelial cells, the alveolar epithelial cells the thymus cells and the spleen lymphocytes in white pulp show positive reactivity (marked by black arrows, bar's unit is μm).
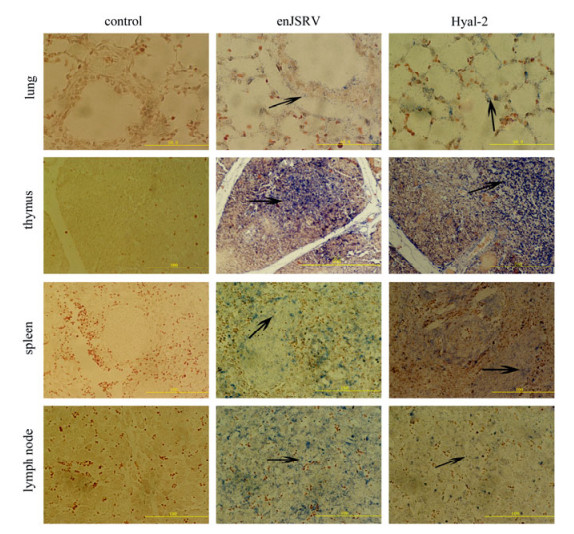
Figure 3. The expression of enJSRV and Hyal-2 in the tissues of 130d fetus by In situ hybridization. Cross sections of the lung, thymus, spleen and mesenteric lymph node from ovine fetus hybridized with DIG-labeled probes. The lung bronchial epithelial cells, the alveolar epithelial cells, the thymus cells, the spleen lymphocytes in white pulp and the lymphocytes in mesenteric lymph node show positive reactivity, (marked with black arrows, bar's unit is μm).
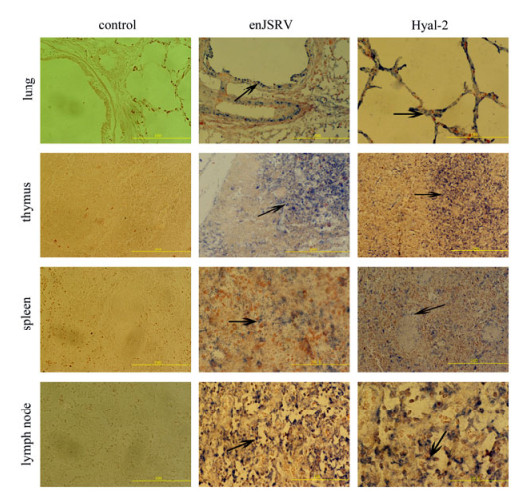
Figure 4. The expression of enJSRV and Hyal-2 in the tissues of 3d newborn lamb by In situ hybridization. Cross sections of the lung, thymus, spleen and mesenteric lymph node from ovine fetus were hybridized with DIG-labeled probes. The lung bronchial epithelial cells, the alveolar epithelial cells, the thymus cells, the spleen lymphocytes in white pulp and the lymphocytes in mesenteric lymph node show positive reactivity (marked with black arrows, bar's unit is μm).

Figure 5. The expression of enJSRV in the tissues of mouse by In situ hybridization. Cross sections of the lung, thymus, spleen from mouse were hybridized with the same DIG-labeled enJSRV probes, all tested tissues did not show any positive signals. (Bar's unit is μm)
-
Real-Time reverse transcription PCR was conducted to analyze the expression of enJSRV and Hyal-2 mRNAs in different tissues (Fig. 6, Fig. 7). Based on the fold changes relative to the lung of 70 d fetuses, the enJSRV mRNA was more abundant by 27-fold in the spleen of 130 d fetuses and 22-fold in the thymus of 3 d newborn lambs. The expression level of enJSRV mRNA in the thymus and mesenteric lymph nodes of 130 d fetuses differed by approximately 10 fold. It is clear that the level of enJSRV mRNA was much higher in immune organs than that in lungs. In addition, the level of enJSRV mRNA was lower in all the tissues of 70 d fetuses than in other periods while there were no significant differences among the expression levels of Hyal-2 mRNA in all collected tissues.
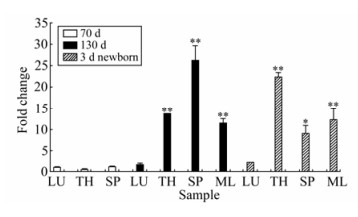
Figure 6. Quantitative analysis of the expression of enJSRV mRNA in different tissues: lung(LU), thymus(TH), spleen (SP) and mesenteric lymph node (ML). The expression level was analyzed by one-way ANOVA followed by Duncan's test. p* < 0.05; p** < 0.01 as compared to the control. The expression of enJSRV mRNA in lung of 70d fetus was considered as control group.
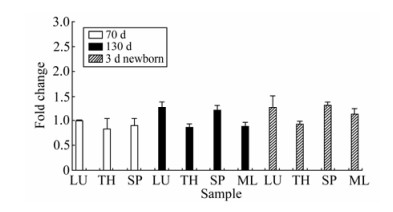
Figure 7. Quantitative analysis of the expression of Hyal-2 mRNA in different tissues: lung(LU), thymus(TH), spleen (SP) and mesenteric lymph node (ML). The expression level was analyzed by one-way ANOVA followed by Duncan's test. The expression of Hyal-2 mRNA in lung of 70d fetus was considered as control group.
Results of RT-PCR
Results of In Situ Hybridization
Results of Real-Time PCR
-
Many experiments have detected the expression of enJSRV mRNA in the placenta and genital tract of mammals[16, 19, 26], but research on the expression of enJSRV mRNA in immune organs of fetuses is limited. In this study, we designed primers and labeled probes according to the hyper-variable region (env region). The expression of enJSRV and Hyal-2 mRNA in fetus′ immune organs was detected by In Situ Hybridization. Thymus, spleen and mesenteric lymph nodes were analyzed because they are crucial for the development of the ovine immune system. The lungs are the primary site of replication of exJSRV. In addition, this is the first report that quantifies the expression of enJSRV and Hyal-2 mRNAs; it may be useful in further studies on the hypothesis that enJSRV expression in the fetus introduces tolerance to the related exogenous viruses[19, 25].
At present, little is known about the relationship between exJSRV and the immune reaction of the host. The etiopathogenesis of OPA and the function of enJSRV in the development of the fetus are still not clear. Nakagawa[11] thought enJSRV did not play a direct role in carcinogenesis, but can induce the body to form immune tolerance. Previous studies indicate that enJSRV can be detected in lung, kidney, thymus, marrow, spleen and mediastinal lymph node using sensitive PCR technology[16]. Spencer[25] reported that the expression of enJSRV mRNA in uterus endomembrane and glandulae uterinae endepidermis of ovine, mesenteric lymph node and thymus of fetus can be detected by In Situ Hybridization.
It is well known that JSRV-infected ovine does not show an appreciable antibody response to JSRV and this leads to the hypothesis that ovines are tolerated towards infection by exJSRV during the embryonic development period. However, the potential immunological mechanism of enJSRV has not been investigated. It is interesting that the results we obtained support this hypothesis well, because we observed a positive signal both in lymphatic and reticular elements located in the medulla of the thymus lobules where negative selection occurs. During the maturity of T cells in the thymus microenvironment, they will experience both positive selection and negative selection processes. The positive selection process will enable the proliferation of T cell clones to produce functional mature T cells, while the negative selection process of T cell clones will make T cell clones eliminated and inform natural immune tolerance[6].
Since enJSRV is expressed in fetus immunologic organs during the embryo period, especially in thymus lobules, it is possible for enISRV to make contact with immature T lymphocytes and then the immature T lymphocytes probably recognize enJSRV as a self antigen and establish a natural tolerance state. However, at the amino acid level, enJSRV is 94 to 95% identical to JSRV in the Gag protein, 95 to 99% identical in the Pro protein, 98% identical in the Pol protein, and 92% identical in the Env protein. It suggests that this high degree of homology make the host immune system recognize exJSRV as a self antigen and therefore not respond to the virus, thereby circulating antibodies cannot be detected in the peripheral blood of JSRV-infected animals.
The results of Real-Time reverse transcription PCR indicated that higher levels of enJSRV mRNA were detected in immune organs, especially in the spleen and the thymus. These data show that enJSRV is strongly localized in the fetus lymphoid tissue. Recently, many studies have shown that thymus grows rapidly and has strong hematopoietic activity during the late embryonic and new-born periods[4]. At the same time, the hematopoietic function of spleen reaches a peak and the number of lymphocytes increases rapidly. This coincides with the results that the level of enJSRV expression is higher during the late embryonic and new-born periods.
The data of Spencer[25] indicated that the cellular receptor for both JSRV and enJSRV is Hyal-2, and enJSRV can interfere with the invasion of exJSRV through two pathways. First, the available receptor for exJSRV was reduced because of the combination of enJSRV and Hyal-2; Second, when enJSRV and exJSRV are expressed in host cells at the same time, virus particles formed by enJSRV cannot be released from the host cell surface, and also prevent the formation of exJSRV virus particles. Palmarini[15]proposed the target cells of exJSRV involved two types of secretary epithelial cells of the lung. Our Real-Time RT-PCR results indicate that the expression of enJSRV is lower in lungs than other tissues. It suggests that there would be more Hyal-2 available as receptor for JSRV in this tissue and is consistent with the evidence that JSRV is able to infect cells in the lung. The high level of enJSRV in immune organs at different periods indicates enJSRV might play an important role in the development of immune organs of fetuses and lambs. On the other hand, previous studies showed super antigens can make the host produce immune tolerance[24]. For example, a mouse mammary tumor virus (MMTV) model can product a protein named endogenous Minor lymphocyte-stimulating (MLs) antigen which is considered as a kind of super antigen. Endogenous interference is well established in the MMTV through the expression of an endogenous super antigen[5, 9]. Some people think Human Immunodeficiency Virus (HIV) also may produce an endogenous super antigen in human beings[22]. In the same manner, we proposed enJSRV may produce a similar kind of super antigen, as enJSRV has much similarity to MMTV; however this needs further experiments.
In summary, the expression of enJSRV and Hyal-2 mRNAs in the fetus immune organs were confirmed further. The high level expression in fetus immune organs obtained in the present study will be useful to unravel the pathogenesis of OPA and offers strong evidence for the immune tolerance hypothesis.

 DownLoad:
DownLoad: