











-
Rice stripe virus (RSV), the type species of the genus Tenuivirus, is a pathogen of the graminaceous plant and is transmitted by the small brown planthopper (Laodelphax striatellus)[4]. RSV has filamentous ribonucleoprotein particles (RNPs) and its genome consists of four single stranded-RNAs that encode seven proteins. The complementary sense of RNA1 encodes a protein of 337 kDa, which was suggested to be an RNA dependent RNA polymerase (RdRp)[18]. RNAs 2–4 are ambisense, each containing two ORFs, one in the 5' half of viral RNA (vRNA), and the other in the 3'half of the viral complementary RNA (vcRNA). RNA2 encodes two proteins, NS2 (22.8 kDa) from the vRNA2 and NSvc2 (94 kDa) from the vcRNA2 (Takahashi et al., 1993). RNA3 encodes NS3 (23.9 kDa) from the vRNA3 and a nucleocapsid protein (CP, 35 kDa) from the vcRNA3[10]. RNA4 encodes a nonstructural disease-specific protein (SP) (20.5 kDa) from the vRNA4 and NSvc4 (32 kDa) from the vcRNA4[9]. Until now, only the functions of the NS3 and NSvc4 proteins encoded by RSV genome have been identified. The NS3 of RSV has been identified as a suppressor of RNA silencing. It could significantly reduce the levels of small interfering RNAs (siRNAs) in silencing cells, and could suppress both local and systemic RNA silencing in the plant[21]. The NSvc4 of RSV has been proved to be a movement protein. This protein, localized predominantly near or within the cell walls, could support the intercellular trafficking of a movement deficient Potato virus X in Nicotiana benthamiana leaves[20].
RSV causes severe disease in rice fields and causes significant yield losses in Asia. It depends on the insect vector for transmission, which therefore plays an essential role in this disease. However, up to now little research has been conducted in to how RSV enters into the insect cells and is transmitted by its insect vectors. Analysis of RSV encoded proteins showed that NSvc2 had amino acids similar to the membrane glycoproteins of some members in the family Bunyaviridae[3]. Bioinformatic proteomic analyses suggested that sequence or structural features of bunyavirus Gc were present in RSV NSvc2. These included a consensus class Ⅱ fusion peptide and a carboxyl terminal transmembrane domain, which were the most highly conserved sequences among type members of five Bunyavirus genuses[7]. The glycol proteins of several members in the family Bunyaviridae, such as bunyamwera virus (BUNV)[14, 15, 16], rift valley fever virus (RVFV)[5], hantaan virus (HTNV)[13] and tomato spotted wilt tospovirus (TSWV)[19], are reported to be class Ⅱ fusion proteins and mediate acidic pH-dependent cell fusion. The fusion peptides of these viruses were found to be critical for inducing membrane fusion. Thus, NSvc2 might also induce membrane fusion in the same way as these glycoproteins.
To provide a more suitable source of NSvc2 to investigate possible protein–membrane interaction, we sought to express NSvc2 on the cell surface in a form analogous to the membrane fusion proteins of enveloped viruses. It has been reported that a display vector containing the gp64 signal peptide and a membrane anchor from the vesicular stomatitis virus (VSV) G glycoprotein showed a high level display of a foreign protein on the surface of virus-infected cells and on the surface of budded virions[1]. This system also proved feasible in studying the membrane fusion activities of several nonenveloped viruses. For example, VP2 of bluetongue virus and the VP2 and P2 capsid protein of rice dwarf phytoreovirus were expressed on the cell surface using this system and confirmed to have membrane fusion activities[6, 22]. In this article, we employed the method previously reported by Chapple and Jones and used a Bac-to-Bac baculovirus expression system to construct a membrane surface display system. Then, we used it to express NSvc2 and CP on the surface of insect cells as glycoproteins to analyze their membrane fusion activities in order to gain an insight into how RSV enters the insect cells.
HTML
-
Spodoptera frugiperda (Sf9) cells were grown in Grace's insect cell culture medium (Invitrogen) supplemented with 10% fetal calf serum (Hyclone). Recombinant viruses were propagated and titered in Sf9 cells.
Baculovirus gp64 AcV1 (a gift from Garry W. Blissard) was a mouse monoclonal antibody raised against extracellular virus of Autographa californica multiple nucleopolyhedrosis virus (AcMNPV). Rabbit polyclonal anti VSV-G-tag (YTDIEMNRLGK, the C-terminal 11 amino acids of vesicular stomatitis virus glycoprotein) antibodies were purchased from Genscript Corp. The secondary antibodies used in immunofluorescence microscopy were fluorescein isothiocyanate (FITC)-conjugated goat anti-rabbit immunoglobulin G (IgG, Beyotime Institute of Biotechnology).
-
The coding region of the gp64 signal peptide was first amplified by PCR using primers Gp-F and Gp-R (Table 1), digested by BamH Ⅰ and Sal Ⅰ, and ligated into the pFastBac1(Invitrogen) to achieve pBac-Sp that encodes the gp64 signal peptide downstream of the polyhedrin promoter. Then, the transmembrane (TM) and cytoplasmic terminal domains(CTD) of the VSV G protein spanning position 4401–4613 bp of the vesicular stomatitis virus (VSV) genome were amplified by PCR using primers VSV-F and VSV-R that introduced Not Ⅰ and Hind Ⅲ sites(Table 1). Following digestion, the PCR fragment was ligated into the plasmid pBac-Sp, to create the plasmid pBac-TM.

Table 1. Primers using in this study
In order to assess the TM domain of the VSV G protein as a basis for baculovirus surface display, the eGFP gene fragment was amplified from pEGFP-N1 with forward primer GFP-F and reverse primer GFP-R (Table 1) and cloned in frame between the baculovirus gp64 signal peptide and the VSV G TM and CTD domains to produce the transfer vector pBac-eGFP-TM (Fig. 1). As a control, the eGFP gene fragment was also cloned into the plasmid pFastBac1 to generate a transfer vector pBac-eGFP that does not contain the gp64 signal peptide and VSV G TM and CTD domains.
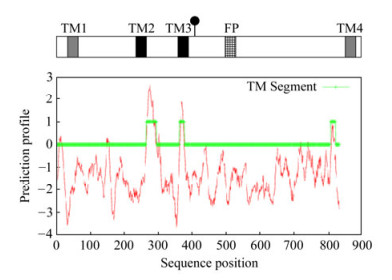
Figure 1. Schematic representation of putative transmembrane helices (TM) and fusion peptide (FP) within the NSvc2. The strongly and weakly predicted transmembrane helices were represented by black and gray squares, respectively. The fusion peptide (FP) was shown as hatched box. The predicted cleavage site was marked with (●). The hydrophobicity plot of NSvc2 is shown at the bottom of the panel.
-
Total RNA was extracted from RSV infected rice leaves with Trizol and used as the template for RT-PCR. Full length (NSvc2) and two mutants (N380 and C383) of NSvc2 gene fragments were amplified with their respective primer pairs (NSvc2-F1/NSvc2-R1 for NSvc2, NSvc2-F1/NSvc2-R2 for N380, and NSvc2-F2/NSvc2-R1 for C383). After restriction digestion with Sal Ⅰ and Not Ⅰ, these amplicons were ligated into pBac-TM to generate the transfer vector pBac-NSvc2-TM, pBac-N380-TM and pBac-C383-TM, respectively. The coding region of RSV CP was amplified with primers CP-F and CP-R and then ligated into pBac-TM to generate pBac-CP-TM using the same method as described above.
A series of constructed transfer vector plasmids were individually transformed into DH10Bac competent cells. The recombinant clones were identified by LacZ selection on the X-Gal and IPTG plates. The recombinant Bacmid DNA were isolated from selected colonies according to the manufacturer's instructions (Invitrogen) and then checked by PCR.
All procedures for the production of viral particles were performed according to the manufacturer's manual (Invitrogen). Briefly, recombinant bacmid DNA in 10 μL Lipofectamine 2000 (Invitrogen) were transfected into Sf9 cells in 35 mm plates. Transfected cells were incubated for 4 h at 27 ℃, and the transfection medium was replaced with fresh medium. After incubation for 96 h at 27 ℃, the recombinant viruses were collected and titered by plaque assay.
-
The recombinant baculoviruses expressing the chimeric proteins were used to infect a monolayer of Sf9 cells at a multiplicity of infection (MOI) of 5 on sterile cover slips (placed in 24-well plates). After 48 h, the infected cells were washed with PBS three times (5 min/each time) and fixed in 4% paraformaldehyde. The cells were then incubated with primary antibodies (polyclonal to VSV-G tag) diluted in PBS containing 1% BSA for 1 h at room temperature. After three washes in PBS, the cells were incubated with FITC-conjugated secondary antibody diluted in PBS containing 1%BSA for 1 h at 37 ℃. The cells were finally washed in PBS, and mounted on glass slides with 50% glycerol. Samples were analyzed on a Zeiss LSM 510 confocal microscope and images were obtained by using LSM 510 image browser software.
-
Monolayer cultures of Sf9 cells were infected at MOI of 5. At 48 h post infection, the cells were washed in Grace's insect cell culture medium and were incubated for 1 h with a monoclonal antibody against gp64 (AcV1) at 1:1000 dilutions. To remove unbound antibody, the cells were washed with medium. The cells were then washed with low-pH buffer (PBS, pH 5.0) and treated with the same buffer for a further 2 min. To restore the normal pH, the cells were washed twice with medium and then incubated in Grace's insect cell culture medium at 28 ℃. Syncytium formation was observed by light microscopy after the pH shift.
Cells, viruses and antibodies
Construction of the transfer vector pBac-TM
Generation of recombinant viruses to express CP, NSvc2 and its derivates on the cell surface
Immunofluorescence
Cell fusion assay
-
Computer analysis of the primary sequence of the RSV NSvc2 protein revealed the presence of four hydrophobic regions (TM1, TM2, TM3, and TM4) as shown in the hydrophilicity profile (Fig. 1). Two of these regions had very high hydrophobicity values (TM2 and TM3), the other two were less hydrophobic.
To identify proteins similar to NSvc2, the protein databases were searched with BLASTX and the results showed that NSvc2 was similar to a membrane protein precursor for phlebovirus members of the Bunyaviridae that is processed into two membrane-spanning glycoproteins G1 and G2. Proteomics computational analyses revealed a collinear arrangement of fusion peptide consensus sequences (YCVTDFHIFSYCPAYHYNM, 484-502 aa) and potential carboxyl terminal transmembrane domains (LVASIIPIGLILKTIRSFL, 809-827 aa), by aligning the carboxyl terminal portion of the NSvc2 protein of RSV with two phlebovirus Gc proteins[7]. The fusion peptide sequence is highly conserved among several Bunyavirus genuses and plays an essential role in cell fusion process[2].
The amino acid sequence of NSvc2 was similar to the membrane glycoproteins of some bunyaviruses and contained a fusion peptide as these glycoproteins, suggesting that it might have similar membrane fusion activity. To test this hypothesis, we investigated the membrane fusion activity of NSvc2 by expressing NSvc2 in insect cells and displaying it on the cell surface.
-
To conveniently study their membrane fusion activities, we proposed to express the proteins on the cell surface as the glycoproteins. The surface display system was generated using Bac-to–Bac baculovirus expression system referred to previously[1]. The gp64 signal peptide and VSV TM and CTD domains were cloned into pFastBacI to generate a baculovirus transfer vector pBac-TM as described in materials and methods.
To test whether the TM region of VSV G protein was the basis for baculovirus surface display, the recombinant baculoviruses Ac-eGFP-VSV and Ac-eGFP(Fig. 2) were generated and then individually infected Sf9 cells. The confocal microscope results for the infected cells showed that the fluorescence of free GFP (from Ac-eGFP) was distributed almost throughout the whole cell, whereas the fluorescence of eGFP-VSV (from Ac-eGFP-VSV) was observed on the surface of the cells (Fig. 3). This indicated that the gp64 signal peptide and the TM and CTD regions of VSV G protein could display the proteins fused to them on the insect cells.
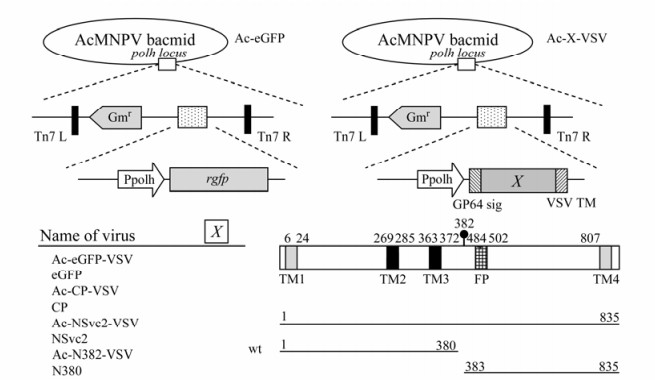
Figure 2. Construction and expression of a series of the membrane-anchored proteins. A baculovirus transfer vector was constructed in which the coding sequences of eGFP, CP, NSvc2 and its derivates were fused in-frame to the signal peptide of the baculovirus gp64 and the C-terminal part of VSV G(right). As a control, the eGFP was expressed directly under the polyhedron promoter (left). The predicted cleavage site is marked with (●) above the position. Abbreviations used are: Gp64 sig, the signal peptide of the AcMNPV major surface antigen gp64; VSV TM, the transmembrane and cytoplasmic domains of the VSV G protein; eGFP, an enhanced fluorescence GFP variant.
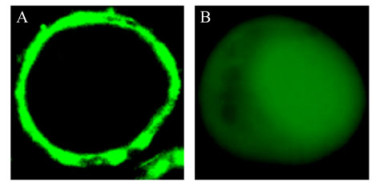
Figure 3. Expression of eGFP in Sf9 cells. Sf9 cells were infected with recombinant baculovirus Ac-eGFP-VSV (A) or Ac-eGFP (B). The cells were visualized on a Zeiss LSM510 confocal microscope.
-
The coding regions of CP, NSvc2, N380 and C383 were individually cloned into pFast-TM and then transformed into DH10Bac to achieve recombinant baculoviruses Ac-CP-VSV, Ac-NSvc2-VSV, Ac-N380-VSV and Ac-C383-VSV, respectively (Fig. 2). To verify that the recombinant proteins were displayed on the cell surface, the cells were cultured on sterile cover slips and infected with recombinant baculovirus at a MOI of 5. At 48h post infection, the infected cells were incubated with primary antibody (rabbit polyclonal anti VSV-G-tag) and then FITC-conjugated secondary antibody. The infected cells were finally subjected to confocal microscopy. The results showed that CP, NSvc2 and its derivates (N380 and C383) all localized within the plasma membrane, demonstrating the anchoring of recombinant proteins on the surface of Sf9 cells (Fig. 4).
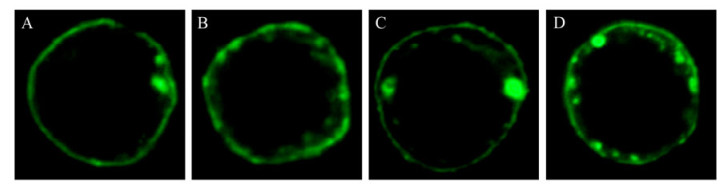
Figure 4. Immunofluorescence assay. Sf9 cells were infected with baculovirus encoding CP (A), NSvc2 (B), N380(C), and C383 (D), respectively. 48 h after infection, the cells were labeled with polyclonal anti-VSV antibodies, followed by FITC-conjugated secondary antibodies, and were visualized on a Zeiss LSM510 confocal microscope.
-
To test the membrane fusion activity at low pH, the cells infected with each recombinant virus were exposed to pH 5.0 for 2 min, followed by incubation at the normal pH 6.2. To confirm that syncytium formation was due to the activity of intended protein but not due to the baculovirus expressed gp64, infected cells were incubated for 1 h with a gp64 monoclonal antibody (AcV1) before pH shift. High numbers of syncytia were detected when these infected cells were shifted to low pH for a short period (Fig. 5 Upper line). In contrast, no syncytium formation observed in infected cells blocked with AcV1 (Fig. 5 Lower line). This demonstrated that the syncytium formation was due to the gp64 but not the recombinant proteins, and none of recombinant proteins (eGFP, CP or NSvc2) expressed in this study has membrane fusion activity on its own (Fig. 5G, H, I).
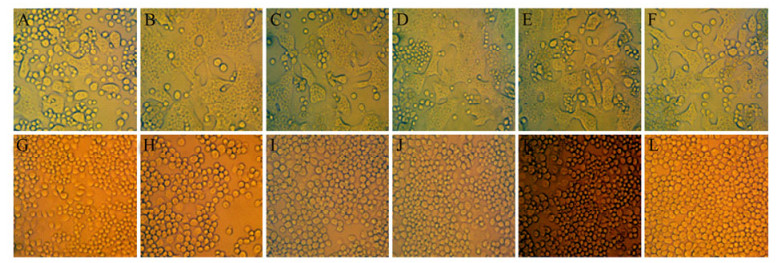
Figure 5. Fusogenic activity of CP and NSvc2. Sf9 cells were infected at a moi of 5, after 48h the infected cells were blocked with AcV1 (low line) or without blocking with AcV1 (upper line), then exposed to pH 5.0 for 2 min, after which the low-pH buffer was replaced by normal growth medium. Pictures were taken on an inverted light microscope after 5h pH shift. Insect cells were infected with one virus Ac-eGFP-VSV (A and G), Ac-CP-VSV (B and H), Ac-NSvc2-VSV (C and I), coinfected with two viruses Ac-N380-VSV and Ac-C383-VSV (D and J), Ac-CP-VSV and Ac-NSvc2-VSV (E and K), and three viruses Ac-CP-VSV, Ac-N380-VSV and Ac-C383-VSV(F and L).
Alignment of RSV NSvc2 with the membrane glycoproteins of phlebovirus showed that a common cleavage site was present in them. Therefore, like the phlebovirus virion membrane glycoproteins, NSvc2 may be post-translationally cleaved and the proposed cleavage site may act as a signal peptide. NSvc2 was predicted to be cleaved into two portions: the N terminus from 1 to 382aa (N380) and the C terminus from 383 to 835aa (C383). The cleavage of NSvc2 possibly did not happen when it was expressed alone in Sf9 cells and this might affect the fusion activity of NSvc2. To test whether it was cleavage that affected the fusion activity of NSvc2, the cells were coinfected with Ac-N380-VSV and Ac-C383-VSV and the syncytium formation triggered by low pH was observed. No syncytium was detected in cells infected with both Ac-N380-VSV and Ac-C383-VSV (Fig. 5J), suggesting that NSvc2 has no fusion activity whether cleaved or not.
Viral entry mechanisms are commonly multistep processes and many viruses use two or more proteins in concert to achieve cell entry. For most viruses, the capsid protein (CP) is usually involved in virus entry. Hence, the CP may have a direct influence on the fusogenic activity of NSvc2. To investigate this possibility, insect cells were infected with a virus expressing either NSvc2 alone or coinfected with viruses expressing both CP and NSvc2 on the cell surface. Additionally, to test whether the NSvc2 cleavage and CP jointly affect the fusion activity, Sf9 cells were coinfected with three recombinant viruses to detect membrane fusion. At 48 h after infection, the infected cells were exposed at pH 5.0 for 2 min and were then shifted back to a normal pH growth medium. Formation of syncytia was monitored by light microscopy after 5h. Consistent with the results obtained with NSvc2-VSV alone, no syncytia could be detected in the cells coinfected with NSvc2 and CP (Fig. 5E, K) or the cells coinfected with CP, N380 and C383(Fig. 5F, L). These results demonstrated that neither NSvc2 nor CP has a specific role in inducing membrane fusion at low pH.
RSV NSvc2 was similar to the phlebovirus viron membrane glycoproteins and contained a fusion peptide
Generation of constructs to express proteins on the cell surface
Expression of NSvc2 and its derivates on the surface of the insect cells
Neither NSvc2 nor CP has membrane fusion activiy
-
Using a baculovirus surface display system, RSV CP, NSvc2 and its truncated proteins were successfully displayed on the insect cell surface. Although the subcellular localization of CP is in the cytoplasm and NSvc2 is in the endoplasmic reticulum in insect cells (data not shown), the surface display system transported all these proteins onto the cell surface. So, this system is a powerful tool to change the native position of target proteins for surface display.
RSV NSvc2 carries stretches of amino acid sequence showing similarity to parts of the membrane glycoproteins of some phleboviruses and contains a fusion peptide that is highly conserved among orthobunyaviruses and which plays an essential role in the membrane fusion process, suggesting NSvc2 possesses membrane fusion activity. However, the results in this study demonstrated that NSvc2 was not able to induce membrane fusion, unlike its homologous glycoproteins. Thus, although RSV NSvc2 has some similarities with the phlebovirus and tospovirus counterparts there are also differences, which could suggest different functions.
Both enveloped and nonenveloped viruses need to deliver their genetic material into host cells for infection. Receptor-mediated endocytosis is the prevalent way for many of these viruses to enter the cell. For enveloped viruses, fusion between viral and cellular membranes results in delivery of the enveloped viral nucleacapsid into the cytoplasm[8], which is mediated by many fusion proteins or their homologues. In contrast, most nonenveloped viruses enter by partially disrupting endosome membranes to release nucleic acids, and this process usually depends on the viral capsid proteins[11]. No enveloped virion has been found in either plants or insects infected by RSV so far, which suggests it is a nonenveloped virus[12]. The data provided here suggests that RSV does not enter the insect cells by way of fusion with the plasma or endosome membrane. RSV might gain entry into cells by forming a transmembrane pore or by partially disrupting endosome membranes, but the mechanism of entering into insect cell still needs further study.

 DownLoad:
DownLoad: