









-
Influenza A viruses contain an eight-segmented, single-stranded, and negative-sense RNA genome and can be differentiated from type B and C influenza viruses based on the internal antigens, the nucleoprotein (NP) and matrix (M1) proteins (Palese P, et al., 2007; Wright P F, et al., 2007). Among influenza A viruses, 16 hemagglutinin (HA) (H1-H16) and 9 neuraminidase (NA) (N1-N9) subtypes have been identified based on major differences within the HA and NA surface glycoproteins(Palese P, et al., 2007; Wright P F, et al., 2007). An influenza pandemic is an epidemic of a possibly emerging virus strain that spreads on a worldwide scale and infects a large proportion of the human population in which individuals might have encountered previous viral strains but have no immunity against these emerging strains(Nicholls H, 2006). Thus, pandemics can cause substantial morbidity and mortality, and pose a significant public health concern(Palese P, et al., 2007; Wright P F, et al., 2007).
Developing better methods for detection of influenza viruses is important to carry out surveillance of virus infection and outbreak in both humans and animals. While the traditional "gold standard" methods include culturing viruses and immunoassays (Amano Y, et al., 2005), the molecular diagnostic tests include reverse transcription-PCR (RT-PCR), microarray, molecular nucleic acid hybridization, quantitative real time RT-PCR (qRT-PCR), mismatch amplification mutation assay (MAMA) PCR, and multiplex RT-PCR assay (Burns N, et al., 1994; Dawson E D, et al., 2007; Hata M, et al., 2007; Storch G A, 2000; Wu C, et al., 2008). As viral strains change and evolve due to constant mutating of the virus genome sequence, this has presented a challenge to develop a universal system or assay that can detect every strain, and poses a need for developing better detection tools in order to monitor the distribution and outbreaks of influenza virus.
In 2009, infection associated with a new swine-origin strain of H1N1 influenza A virus (called pandemic H1N1/2009 strain herein) was declared to be a global pandemic (Stage 6) by the World Health Organization (WHO) after evidence of spreading in the world(Garten R J, et al., 2009; Vijaykrishna D, et al., 2010). By the end of 2009, more than 200 countries had officially reported over more than 500, 000 laboratory confirmed cases of the influenza pandemic H1N1/2009 infection, including more than 6, 250 deaths(World Health Organization, 2009). In China, there were more than 123, 000 laboratory confirmed cases, including at least 700 deaths by the end of 2009(Cao B, et al., 2009). The incidents associated with the H1N1/2009 influenza virus had declined significantly since early 2010, leading to the WHO announcement of the end of the epidemic in late 2010 (World Health Organization, 2010). However, it is unclear whether outbreaks associated with this novel strain will occur again, and sporadic cases of infections associated with this strain and its derivative strains have been found in China in 2011 (Yang, Z., Mao, G., Liu, Y., Liu, W., Luo, Y., Wu, J., and Liu, F., unpublished results). Today, it is still important to continue developing better and improved assays that can rapidly and specifically detect the pandemic H1N1/2009 influenza virus as well as its derivative strains in order to monitor the infection of these viruses in the human and animal populations.
As a rapid response to the outbreak of the pandemic H1N1/2009 virus, the WHO provided a quantitative real time RT-PCR (qRT-PCR)-based assay for detection of this virus in October 2009 (World Health Organization, 2009). The WHO-recommended method included a panel of oligonucleotide primers and dual-labeled hydrolysis (Taqman®) probes to be used in qRT-PCR assays for the in vitro detection and characterization of the novel H1N1/2009 strain in respiratory specimens and viral cultures. The primers and probes targeted the conserved regions of the HA genes of the pandemic viruses. In addition, many assays that were based on the qRT-PCR technology or the WHO-recommended systems have been developed and studied (Bennett S, et al., 2010; Gunson R, et al., 2010; Jiang T, et al., 2010; Klungthong C, et al., 2010; Lorusso A, et al., 2010; Pabbaraju K, et al., 2009; Schulze M, et al., 2010; Yang Y, et al., 2011). These assays using the oligonucleotides have greatly facilitated the detection and monitoring of the infections associated with the pandemic H1N1/2009 virus.
As influenza viruses spread and replicate, mutations are constantly introduced into the viral genome(Palese P, et al., 2007; Wright P F, et al., 2007). Thus, potential mismatch of the sequences in the emerging strains with the primers and probes of the WHO-recommended assay arises, and may reduce the sensitivity and effectiveness of the WHO assay for detecting the pandemic H1N1/2009 strain (Klungthong C, et al., 2010; Pabbaraju K, et al., 2009; World Health Organization, 2009). It is important to continue developing better and improved assays, including new sets of primers and probes with optimal match of virus sequences that can be used in the qRT-PCR assays, for detection and surveillance of the infections associated with the pandemic H1N1/2009 strains.
In this study, we have developed a novel set of primers that can be used in a qRT-PCR assay for detection of the pandemic H1N1/2009 influenza virus. A primer-probe set recommended by WHO was used as a reference method. The newly designed primers target three regions that are highly conserved among the HA genes of the pandemic H1N1/2009 viruses and are different from those targeted by the WHO-recommended primers. The qRT-PCR assays with the newly designed primers are highly specific in detecting pandemic H1N1/2009 viruses. Furthermore, the qRT-PCR assays with our designed primers appeared to be 10-fold more sensitive than those with the WHO-recommended primers, with a detection limit of about 2.5 copies of target RNA per reaction. Of 83 human clinical nasopharyngeal swabs tested, 32 were detected to be positive using the qRT-PCR assays with the newly designed primers, while only 25 were positive in the assays with the WHO-recommended primers. These results suggest that the qRT-PCR assay with the newly designed primers is highly sensitive for detecting the pandemic H1N1/2009 virus infection. Furthermore, our study demonstrates the feasibility of developing highly sensitive detection assays for early diagnosis of emerging influenza virus strains.
HTML
-
All research involving human participants was approved by the Institutional Review Boards of the Taizhou Institute of Virology and College of Life Sciences, Wuhan University, in accordance with the guidelines for the protection of human subjects. Written informed consent was obtained from each participant. No research involving human participants was carried out at the University of California-Berkeley.
-
A total of 83 clinical samples of nasopharyngeal swab were collected at the People's Hospital of Taizhou, China from December 5, 2009 to January 10, 2010. The H1N1/2009 reference strain (A/Taizhou/09/2009 (H1N1)) was isolated from a clinical sample of nasopharyngeal swabs obtained from the People's Hospital of Taizhou. The genomic segment coding for the HA gene was cloned and its sequence was determined(accession number JN863077). The other virus strains used in this study (Table 1) have been described previously (Gu H, et al., 2010; Li X, et al., 2009; Qi X, et al., 2009). All the virus isolates were grown in chicken eggs and the infectious allantoic fluids were collected and used as the virus stocks and samples. The titers of the virus samples were determined by plaque assays in Madin-Darby canine kidney (MDCK) cells using a standard protocol (Salomon N, et al., 1999). Total RNA samples were isolated from 140 μL samples of the allantoic fluid or nasopharyngeal swabs (suspended in Dulbecco's modified Eagle medium, DMEM) using the QIAamp viral RNA mini extraction kit (QIAGEN, Shanghai, China) in accordance with the manufacturer's protocol, and were stored at -70℃ until used.

Table 1. Virus isolates used in this study
-
The HA genomic segment of the reference strain (A/Taizhou/09/2009 (H1N1)) was cloned following the procedures described previously (Gu H, et al., 2010; Li X, et al., 2009; Qi X, et al., 2009). Briefly, RNA was extracted from the reference strain using Trizol reagent (Invitrogen, Shanghai, China). The HA sequence was reverse transcribed into cDNA and amplified by PCR with the uni12 primer and specific primer sets described previously (Hoffmann E, et al., 2001). The PCR-amplified products were cloned into vector pMD-18T (TAKARA, Dalian, China) and sequenced to verify its accuracy (Invitrogen, Shanghai, China). The resulting plasmid, designated as pT-HA (H1N1/2009/Taizhou), was used as the template for the subsequent studies.
-
The qRT-PCR primers and probes were designed based on the sequence information obtained from the Influenza Sequence Database (http://www.ncbi.nlm.nih.gov/genomes/FLU/FLU.html). Each of the available pandemic H1N1/ 2009 virus HA sequences from the database were aligned, and primers and probes were designed specifically to target the highly conserved regions of these sequences using the Beacon Designer (version7.6) program (Premier Biosoft). The primers and probes that were used in the study and yielded the best qRT-PCR results in detecting the pandemic H1N1/2009 virus were listed in Table 2.

Table 2. Primers and probes used in our developed qRT-PCR assays for the detection of the pandemic H1N1/2009 virus. A primer-probe set recommended by WHO, which was used as a reference method in our study, is also included
-
The RNA transcript corresponding to the HA gene of a reference strain (A/Taizhou/09/2009 (H1N1)) was in vitro transcribed from construct pT-HA (H1N1/2009/Taizhou) and used as the template to optimize qRT-PCR conditions. The qRT-PCR reactions were optimized at different temperatures and times, using the reagents and procedures as described in the WHO-recommended qRT-PCR assays (World Health Organization, 2009). The qRT-PCR reactions were performed in a reaction mixture containing 1 μL in vitro transcribed RNA of HA (H1N1/2009/Taizhou), 12.5μL 2X reaction mix and 0.5μL SuperScriptTMⅢ RT/Platimun® Taq Mix (SuperScriptTMⅢ Platinum® One-Step Quantitative Kit (Invitrogen, Ct.no.11732-020), Invitrogen, Shanghai, China), and 800 nmol/L of the forward and reverse primers and 200 nmol/L of Taqman probe that were either recommended by the WHO or generated based on our primer design. The reaction was performed for 30 min at 50℃ followed by 2 min at 95℃ with subsequent 45 cycles of amplification (95℃ for 15 s and 55℃ for 30 s), and the fluorescence was recorded at 55℃. For determination of the optimal melting temperature, the melting temperature was set at 55.0, 56.1 57.0, 58.3, 59.2, and 60.0℃. Regardless of the WHO-recommended or the newly designed primers to be used, the qRT-PCR reactions consistently yielded the lowest threshold cycle (Ct) values at a melting temperature of 55℃. Accordingly, the qRT-PCR assays described in the study were carried out under these conditions unless specified.
-
RNAs were in vitro synthesized by T7 polymerase from constructs containing different gene segment (HA, NA, PA, PB1, PB2, NP, M, NS) of a human seasonal H1N1 influenza virus reference isolate (A/Nanchang/ 14/1996) and the HA sequences of other viruses listed in Table 1. These constructs have been either constructed in our laboratory or described previously (Gu H, et al., 2010; Li X, et al., 2009; Qi X, et al., 2009). The in vitro transcribed RNAs were used as the templates to assess the specificity of the qRT-PCR assays with the designed primers. All the experiments were performed in triplicate and repeated three times.
To study the sensitivity of the qRT-PCR assays, total RNA was extracted from infectious allantoic fluid containing the reference virus strain (A/Taizhou/09/2009 (H1N1)) using QIAamp viral RNA mini kit (QIAGEN, Shanghai, China). Serially diluted RNAs ranging from 10-1 to 10-6 were used in the sensitivity test and the detection limit of the qRT-PCR assay was determined.
To further determine the sensitivity of the assays, RNA was in vitro transcribed from construct pT-HA (H1N1/ 2009/Taizhou) using the RiboMAXTM Large Scale RNA Production System-T7 (Promega, Beijing, China). Serial dilutions of the in vitro transcribed RNAs, which range from 2.5 to 5×106 copies per reaction, were used as the template for the sensitivity test.
The qRT-PCR reaction was performed following the WHO recommendations (World Health Organization, 2009). The assays were carried out, using the SuperScriptTMⅢ Platinum® One-Step Quantitative Kit (Invitrogen), in a 25 μL mixture containing 5 μL of serial diluted RNA samples, 12.5 μL 2X Reaction mix, 0.5μL SuperScriptTMⅢ RT/Platimun® Taq Mix (Invitrogen), and 800 nmol/L of forward and reverse primers and 200 nmol/L of Taqman probe that were either recommended by the WHO or generated based on our primer design. Amplification and detection were performed on a Bio-Rad iCycler iQ5TM real time PCR system (Hercules, CA) for 30 min at 50℃ followed by 2 min at 95℃ with subsequent 45 cycles of amplification (95℃ for 15 s and 55℃ for 30 s), and the fluorescence was recorded at 55℃. All the experiments were done in triplicate and repeated three times.
Ethics Statement
Virus strains and culture, and clinical samples
Construction of the plasmid containing the HA sequence of the H1N1/2009 virus
Primer design
Optimization of qRT-PCR reaction conditions
Specificity and sensitivity studies
-
To design the qRT-PCR primers, a large number of the sequences coding for the HA gene of influenza viruses that are currently available in the Influenza Virus Resource of National Center for Biotechnology Information (NCBI) (between 1998 and 2010), including the seven strains used in this study (Table 1), were analyzed with the Lasergene DNASTAR software package (DNASTAR Inc, Madison). We searched for regions that were highly conserved among the pandemic H1N1/2009 viruses but had low sequence conservation between other influenza virus strains. Accordingly, qRT-PCR primers were designed to specifically target these regions with the Beacon Designer program (version 7.6) (Fig. 1 and Table 2). Altogether, a primer set consisting of a forward primer, a reverse primer, and a fluorescent hybridizing probe recognizing three distinct and highly conserved regions on the target sequence was generated and used in this study (Fig. 1 and Table 2). A primer-probe set recommended by WHO (Table 2) (World Health Organization, 2009), which targets the HA sequence of the pandemic H1N1/ 2009 viruses, was also included and used as a reference method in our study.

Figure 1. Alignment of sequences of HA genes of various H1N1/2009 virus strains with both a newly designed set of primers and probes and a primer-probe set recommended by WHO. The HA genes were from some of the pandemic H1N1/2009 viruses in Genbank, including the swine H1N1 strain (A/Swine/Shanghai/2005(H1N1)) and the reference H1N1/2009 strain (A/Taizhou/ 09/2009 (H1N1)) used in the study
-
In the WHO-recommended qRT-PCR system, the assay was carried out with a specific set of primers under conditions that have been determined to be optimal (Pabbaraju K, et al., 2009; World Health Organization, 2009). To examine the performance of the generated primers for detection of pandemic H1N1/2009 virus, a series of experiments withdifferent conditions, including the qRT-PCR conditions recommended by the WHO, were conducted. The qRT-PCR assay was performed using construct pT-HA (H1N1/2009/Taizhou), which contained the sequence coding for the HA gene of a reference strain (A/Taizhou/09/2009(H1N1)) isolated in our laboratory, as the template to determine the optimal temperature and time for the reaction. The threshold cycle (Ct) values for the qRT-PCR reactions were determined under each condition, and those conditions under which the qRT-PCR reactions exhibited the lowest threshold cycle (Ct) values were used in the subsequent experiments. For example, qRT-PCR with the designed primer set was carried out at gradient melting temperature 55.0, 56.1 57.0, 58.3, 59.2, and 60.0℃, respectively. Our results indicated that the qRT-PCR using our designed primers performed optimally under the WHO-recommended qRT-PCR conditions with a melting temperature of 55.0℃ as the lowest threshold cycle (Ct) values were observed at this temperature. Similar results were also observed when the qRT-PCR assays were carried out using RNA samples isolated from the reference strain (A/Taizhou/09/2009(H1N1)) as the template (data not shown). The qRT-PCR assays using the primer set recommended by the WHO also exhibited the lowest Ct values under these conditions (Pabbaraju K, et al., 2009; World Health Organization, 2009). Hence, all subsequent qRT-PCR assays were carried out under these optimized conditions.
-
Three sets of experiments were carried out to evaluate the specificity of the developed qRT-PCR assay. First, we searched the sequence homology of the designed primers against all the sequences in the NCBI database. The search results indicated that only the HA genes from the pandemic H1N1/2009 influenza viruses exhibited significant sequence homology with the primers. Second, the specificity of the qRT-PCR assays with the designed primers was evaluated by amplifying the RNAs transcribed from the constructs containing the sequence coding for the eight individual genes (HA, NA, PB2, PB1, PA, NP, M and NS) of a human seasonal H1N1 influenza virus reference isolate (A/Nanchang/14/1996) (Gu H, et al., 2010; Li X, et al., 2009; Qi X, et al., 2009). Positive amplifications were not observed in reactions with these genes as templates.
Third, the specificity of the qRT-PCR assay with the newly designed primers was further evaluated with plasmids containing HA sequences of other subtypes of influenza viruses (e.g. H1, H3, and H5) or an influenza B virus isolate (i.e. H1N1 A/Nanchang/14/1996, H 1N1 A/ swine/Shanghai/2005, H 3N2 A/Swine/Shandong/1/2004, H 5N1 A/Raccoon dog/Shangdong/sd1/2005, and B/ Shanghai/361/2002) (Table 1) (Gu H, et al., 2010; Li X, et al., 2009; Qi X, et al., 2009). In comparison, qRT-PCR assays with the WHO-recommended primers were also carried out in parallel. Positive amplifications were only observed in the qRT-PCR with our designed primers using the HA templates derived from the pandemic influenza virus A/Taizhou/09/2009 (H1N1) and swine H1N1 virus A/Swine/Shanghai/2005 (H1N1) (Fig. 2A). In contrast, the qRT-PCR assay did not amplify the templates from human seasonal H1N1 influenza, and other non-H1 influenza A (e.g. H3 and H5 subtype) or B viruses. Similarly, the qRT-PCR assays using the WHO-recommended primers were able to amplify the sequences of pandemic virus A/Taizhou/09/2009 (H1N1) and swine H1N1 virus A/Swine/Shanghai/2005 (H1N1) but not those of human seasonal H1N1 influenza, and other non-H1 influenza A (e.g. H3 and H5 subtype) or B viruses (Fig. 2B, Table 1). These results suggested that the qRT-PCR assay with our designed primers is specific and is comparable to the specificity of the assay with the WHO primers. Of all the sequences tested, the sequence of the HA gene from A/Swine/Shanghai/2005 (H1N1) had the highest homology to that from the pandemic 2009/H1N1 virus (Fig. 1, see also Fig. 5, data not shown), possibly providing an explanation of the cross-reactivity of the qRT-PCR with both our and WHO-recommended primers to this virus isolate. Similar results were obtained when the assays were carried out in three separate and independent experiments, suggesting that the assays were reproducible experimentally.
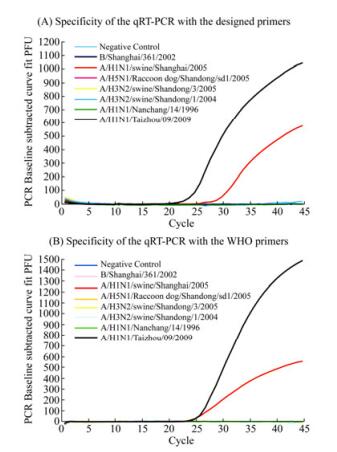
Figure 2. Specificity of the newly developed qRT-PCR assay for detection of the pandemic H1N1/2009 influenza virus. Positive results were observed in the qRT-PCR reactions with the HA sequence from the swine H1N1 strain (A/Swine/Shanghai/2005 (H1N1)) (red) and reference pandemic H1N1/2009 virus (A/Taizhou/09/2009 (H1N1)) (black) using our developed primers (A) and the WHO recommended primers used in our study (B)
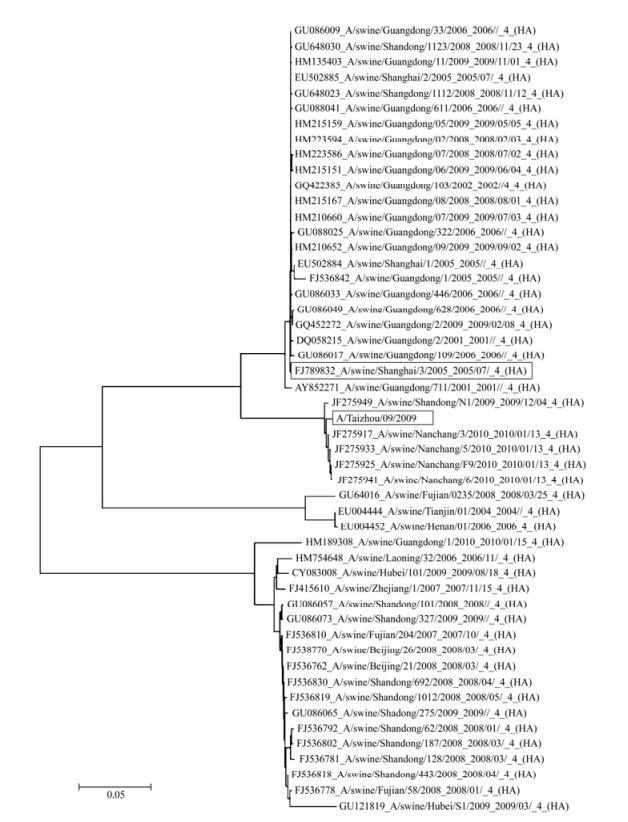
Figure 5. Phylogenetic analysis of the swine-origin H1N1 influenza viruses and the pandemic H1N1/2009 influenza virus strains. The strains in boxes represent the swine H1N1 strain (A/Swine/Shanghai/2005(H1N1)) and the reference pandemic H1N1/2009 (A/Taizhou/09/2009 (H1N1)) strain used in the study
-
To study the sensitivity and detection limit of the methods, the qRT-PCR assays with our designed primers and the WHO recommended primers were carried out in parallel using 10-fold serial dilutions of in vitro transcribed RNAs (ranging from 2.5 to 2.5×106 copies per reaction) coding for the HA gene of the reference strain (A/Taizhou /09/2009(H1N1)) as the template (Fig. 3). Similar results were obtained when the assays were performed in three separate and independent experiments, suggesting that the assays were reproducible experimentally. The sensitivity of the assays with our designed primers and the WHO primers were determined and compared using the same template at identical concentrations. The threshold cycle (Ct) values in the reactions with our designed primers were consistently lower than those with the WHO primers at the same dilution of the in vitro-transcribed RNA template (Fig. 4A). To further confirm these results, the qRT-PCR assays with both the WHO primers and our designed primers were carried out by assaying RNAs extracted from the reference pandemic H1N1/2009 virus (A/Taizhou/09/2009 (H1N1)) of known plaque-forming unit (PFU). RNAs were serially diluted 10-fold and used as the templates. Similarly, the threshold cycle (Ct) values in the reactions with our primers were consistently lower than those with the WHO primers at the same dilution of the RNAs extracted from the virus samples (Fig. 4B). For example, at a 1×105 fold dilution of the samples, the threshold cycle (Ct) value in the reactions with our primers was 32 while that with the WHO primers was 40 (Fig. 4B). The detection limit of the qRT-PCR assay with our primers and with the WHO recommended primers was about 2.5 and 25 copies in vitro-transcribed RNA per reaction, respectively (Fig. 3 and 4). These results indicated that the detection sensitivity of the qRT-PCR assay with our designed primers is about 10-fold better than that of the assay with the WHO-recommended primers.
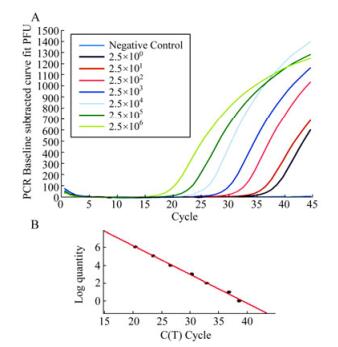
Figure 3. Representative amplification (A) and standard curves (B) in the newly developed qRT-PCR assay with serially diluted-in vitro transcribed RNA of the HA gene. (A). Tenfold serial dilution of the template RNA starting at 2.5x106 copies per reaction was tested. In (B), Ct values calculated from results in (A) were plotted against the log of the initial starting quantity of RNA transcript (copies/uL). The threshold is -0.32, slope is -3.121, R2 is 0.996, E is 109.1%, respectively
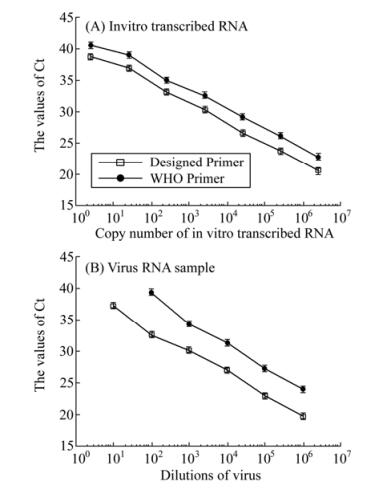
Figure 4. Sensitivity of qRT-PCR assays for the detection of the HA RNA sequence from reference H1N1/2009 virus (A/Taizhou/09/2009 (H1N1)), using the newly developed primer-probe set and a primer-probe set recommended by WHO. The templates used for the qRT-PCR assays were the serially diluted RNA samples that were either directly isolated from reference H1N1/2009 virus (B) or were in vitro synthesized from construct pT-HA (H1N1/2009/Taizhou) containing the HA sequence of the reference H1N1/2009 virus (A), respectively. The qRT-PCR assays were carried out in triplicate and repeated three times. The threshold cycle (Ct) values were the averages of the three independent experiments. The standard deviation is indicated in the error bars
-
To further assess the utility of the developed H1N1 qRT-PCR assay in clinical diagnostics, a panel of 83 human specimens of nasopharyngeal swabs collected between December 2009 and January 2010 was tested. To verify the results independently, the extracted RNA samples were assayed separately and in parallel by the qRT-PCR assays using the design primers and the WHO-recommended primers, respectively. For some of the samples showing positive results in the assays, virus isolation in embryonated chicken eggs or in MDCK cells was subsequently performed and the isolated viruses were characterized.
Of the 83 clinical samples, which had never been tested for influenza viruses prior to the study, 32 were positive for the presence of pandemic H1N1/2009 influenza virus in the qRT-PCR assay with our designed primers (Table S1 and Fig. 6). In contrast, only 25 of these 32 samples were positive in the assay with the WHO-recommended primers. Further isolation of the H1N1/2009 virus from these seven samples that were positive in the assays with our primers but not with the WHO primers confirmed the presence of the influenza virus. Moreover, the HA coding sequences from all these seven samples were cloned and sequencing analyses of these cloned sequences revealed that they were H1N1/2009 virus (data not shown). The inability of the assays with the WHO-recommended primers to detect the seven samples that were positive in the assays with our primers was possibly because the qRT-PCR reaction with the WHO primers is about 10 fold less sensitive than that with our primers.
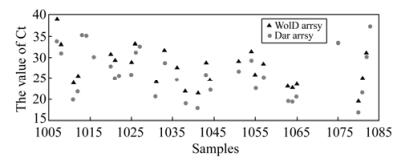
Figure 6. Detection (the value of threshold cycle (Ct)) of the pandemic H1N1/2009 virus in 83 samples by qRT-PCR using our designed primers and the WHO recommended ones. The results were also listed in Table S1
Design of the qRT-PCR primers for pandemic H1N1/ 2009 virus
Optimization of the qRT-PCR condition
Studies of the specificity of the H1N1/2009 qRT-PCR assay
Studies of the sensitivity of the H1N1/2009 qRT-PCR assay
Validation of the developed H1N1 qRT-PCR assay with clinical samples
-
In this study, we report a rapid, sensitive, and specific diagnostic system based on quantitative real-time RT-PCR (qRT-PCR) for the detection of the pandemic H1N1/2009 influenza virus. In this qRT-PCR system, three primers were designed to target a highly conserved HA sequence of the H1N1/2009 virus. Our results indicate that the system accurately and specifically detected pandemic H1N1/2009 virus from cell cultures and human nasopharyngeal swab samples. Importantly, the qRT-PCR assay using our newly designed primers can detect as little as 2.5 copies of viral RNA sequence per reaction. Thus, the qRT-PCR system developed in this study is highly sensitive for detecting pandemic H1N1/2009 influenza virus.
Because of the high mutation rate of the influenza virus genome sequence, including the HA gene segment, it is difficult to generate a single set of qRT-PCR primers to detect every isolate of the pandemic H1N1/2009 influenza virus (Bennett S, et al., 2010; Gunson R, et al., 2010; Jiang T, et al., 2010; Klungthong C, et al., 2010; Lorusso A, et al., 2010; Pabbaraju K, et al., 2009; Schulze M, et al., 2010). We have carefully analyzed most of the HA gene segments of pandemic H1N1/2009 influenza virus available in the Influenza Virus Resource of National Center for Biotechnology Information (NCBI) and designed a set of novel primers specifically targeting three regions of HA gene segments that are highly conserved among pandemic H1N1/2009 influenza strains. These three regions are distinct from those targeted by the WHO-recommended primer-probe set used in our qRT-PCR assays (Fig. 1). The developed qRT-PCR assay based on our newly designed primers is highly accurate and specific for detecting each of the viral strains tested (Table 1), even though the primers were designed based on the general features of the HA sequences of the pandemic H1N1/2009 viruses available in the Influenza Virus Resource and were not specifically based on the sequences of the viral isolates used in this study. These results further suggest that our design strategy of qRT-PCR primers and probes should be useful to construct novel primers for detection of other influenza strains in China and in other parts of the world.
In the WHO-recommended qRT-PCR system, the assay was carried out with a specific set of primers under conditions that have been determined to be optimal (Pabbaraju K, et al., 2009; World Health Organization, 2009). Our results suggest that the developed qRT-PCR system with our primers worked optimally under these conditions and exhibited high specificity and sensitivity when assayed with 7 different virus isolates of swine, human, and raccoon dog origin, and with 83 clinical samples of human nasopharyngeal swabs. The developed system only amplified and detected the HA gene segments of pandemic H1N1/2009 influenza virus and a swine H1N1 virus strain which HA gene sequence is highly conserved with that of pandemic H1N1/2009 influenza virus (Fig. 1). In order to analyze the relationship between this swine influenza virus strain and the pandemic H1N1/2009 influenza virus, we have carried out a phylogenetic analysis of this swine virus and the 2009 H1N1 viruses (Fig. 5). Our analysis suggested that the evolutionary relationship between this swine influenza virus strain and the pandemic 2009 H1N1 viruses (e.g. A/Taizhou/09/2009 (H1N1)) is closer than other swine virus strains. Further validation of the specificity of the developed qRT-PCR assay with our primers using each segment (HA, NA, PA, PB1, PB2, NP, M, NS) of a human seasonal H1N1influenza virus referenceisolate (A/Nanchang/14/1996) and the HA sequences of other subtype (e.g. H3 and H5) isolates and human samples revealed that there was no cross-reactivity with influenza virus other than the HA sequence of the pandemic H1N1/2009 viruses (Fig. 2). Thus, under the optimized assay conditions recommended by the WHO, the specificity of the qRT-PCR assay with our newly designed primers is high and is comparable to that of the assay with the WHO-recommended primers in detecting the pandemic H1N1/2009 virus strains.
Sensitivity is a critical parameter for a diagnostic tool (Gu H, et al., 2010; Pabbaraju K, et al., 2009; Spackman E, et al., 2002). Our results indicated that the detection limit of the qRT-PCR assay with our primers is 2.5 copies of the in vitro transcribed HA RNA per reaction, and is 10-fold lower than that of the assay with the WHO primers, which is 25 copies of in vitro transcribed RNA per reaction. These results suggest that the developed qRT-PCR assay with our primers is 10-fold more sensitive than that with the WHO primers that were used as the reference method in our study.
The qRT-PCR assay with the WHO-recommended primers is a well designed and effective molecular diagnostic test that was successfully deployed worldwide during the pandemic H1N1/2009 influenza virus outbreak (Pabbaraju K, et al., 2009; World Health Organization, 2009). However, the HA sequence diversity that commonly occurs in influenza viruses in different geographical regions and/or time periods resulted in mismatch of the templates with the primers and probes. Mismatches were initially observed aligning primer and probe sequences with 105 randomly selected HA gene sequences of the pandemic H1N1/2009 influenza virus collected mostly from America and Europe during April to July 2009 (Klungthong C, et al., 2010). For example, previous analyses revealed 1-2 mismatches in the probe region, 1 mismatch in the forward primer, and 1-3 mismatches in the reverse primer region (Fig. 1). These mismatches potentially affect the properties of the WHO qRT-PCR assay such as the sensitivity and specificity (Klungthong C, et al., 2010; Yang Y, et al., 2011). Our analyses of the WHO primers that were used as the reference method in our study suggest that one mismatch was found in the probe region at the 16th nucleotide (G to A), and three mismatches in the reverse primer region at the 12th nucleotide (A to G), the 15th nucleotide (G to A), and the 19th nucleotide (A to G), respectively (Fig. 1). In contrast, no mismatch was identified between our designed primers and the published HA sequences of the pandemic virus strains (Fig. 1). It is conceivable that the mismatches associated with the WHO-recommended primers but not with our designed primers may contribute to the difference in the sensitivity of our developed qRT-PCR assays and the WHO assays. Further studies will provide insight into how the sensitivity of qRT-PCR assay for the detection of influenza virus is improved using designed primers with specific mismatch and match with the target sequence.
In this study, we used the qRT-PCR methods with both the WHO-recommended primers and our designed primers to assay clinical samples in parallel. Of 83 human nasopharyngeal swabs, 25 were positive in both assays. Furthermore, seven additional samples were positive only in the reactions with our primers. The H1N1/2009 viruses were successfully isolated from these seven samples. Moreover, the HA coding sequences from all these seven samples were cloned and sequencing analyses of these cloned sequences revealed that they were H1N1/2009 virus, indicating that they represent true positives. The reason that the seven samples were missed by the reactions with the WHO primers was possibly because the sensitivity of the WHO-recommended assay is at least 10-fold lower than that of the developed assay, as our results showed. We note that all the samples that were negative in the assays using our developed primers were also negative in those using the WHO-recommended primers (Table 3 and Fig. 6). Thus, we believe that the developed qRT-PCR assay with our primers is specific and may not yield any false negatives or false positives.
We note that the numbers of our reference isolates and clinical samples in the study were relatively small, and only included several of the known subtypes of influenza virus (i.e. H1, H3, H5 and B). Since the assay was developed against a limited number of cultured isolates and clinical samples, further validation with additional influenza isolates and testing of more clinical specimens is needed to better define the usefulness and effectiveness of the developed qRT-PCR detection system (Bennett S, et al., 2010; Gunson R, et al., 2010; Jiang T, et al., 2010; Klungthong C, et al., 2010; Lorusso A, et al., 2010; Pabbaraju K, et al., 2009; Schulze M, et al., 2010). We believe that our current design for the primers and probes that detect the pandemic H1N1/2009 influenza virus should be easily applied to construct panels of primers for detecting all other influenza virus strains. Assays based on technologies other than qRT-PCR may also be developed for detection of the pandemic H1N1/2009 virus (Chiu C Y, et al., 2008; Ghedin E, et al., 2011; Yongfeng H, et al., 2011). The development of the qRT-PCR technology as well as other approaches should provide powerful tools for the surveillance of influenza viruses in animals and humans in order to prevent and control the diseases associated with these viruses.
-
YZ, GM, YL, KZ, YP, JW, and FL conceived the study, analyzed the results, and wrote the paper. YZ, GM, YL, YC, CL, JL, and XL performed the research. All authors read and approved the final manuscript.
-
We are indebted to Hao Gong and Wang Yu for technical assistance and Paul Rider, Hongwei Gu and Xian Qi for suggestions and reagents.
-
is linked to the online version of the paper on the Virologica Sinica website.www.virosin.org

 DownLoad:
DownLoad: