









HTML
-
Nucleotide and protein sequences of isolates collected from infected populations can be useful for determining the threats, such as host adaptation, which are associated with the emergence of new lineages. March 2013 saw the first reports of a new H7N9 lineage in Shanghai, China that saw a gradual increase in the number of cases; but, by 17th May 2013, four Chinese provinces had ended their emergency response to H7N9 bird flu. However, despite detailed reports on human cases, the publicly available sequences from collected isolates were relatively few. By analyzing the data at this time we show that limited insight can be obtained from a relatively small dataset and, in particular, more avian isolates are necessary to consider host adaption. With reports of new H7N9 cases in South China our findings are relevant to establishing effective monitoring and determining the risk associated with these new events.
The first cases of H7N9 in humans were reported in Shanghai city and Anhui province in March 2013. Subsequent cases were reported in neighboring provinces, consistent with a gradual geographical dispersion of the virus. By the time Jiangsu, Jiangxi, Zhejiang, Anhui provinces and Shanghai city had ended their emergency response, a total of 130 human cases had been reported in mainland China, of which 37 were fatal (Zhu E Z, 2013). In recognition of international concerns regarding the potential threat from this outbreak, the Chinese government released detailed information about all human cases as well as location of bird farms where widespread culling was implemented. As a consequence, it was possible to build up a detailed view of the dissemination and impact of the virus. It appeared that all of the human cases were associated with bird-human transmission and, despite the concerns voiced (Sebelius K, 2013) by various public health bodies of the possibility of host adaptation, there was no evidence that any cases were a consequence of humanhuman transmission.
In addition, various research groups published several sequences from human cases and by the time the end of the emergency response was declared, there were 20 (13 human, 5 avian and 2 environmental) H7N9 sequences deposited in the GISAID database (http://www.gisaid.org). Several published reports attempted to classify a selection of these isolates in terms of their relationship to other related strains, e.g., (Chen Y, et al., 2013; Gao R, et al., 2013; Liu D, et al., 2013; Liu Q, et al., 2013; Xiong C, et al., 2013; Zhang W, et al., 2013), made estimates of mutation rates and considered mutations that could affect pathogenicity and host adaptation.
When a virus undergoes adaptation to a new host, in addition to amino acid variation, a higher nucleotide evolution rate is commonly observed. Early reports (Xiong C, et al., 2013; Zhu E Z, 2013) provided estimates of these rates and suggested they were consistent with other avian influenza outbreaks but, notably, did not provide the associated confidence intervals. When we estimated evolution rates for the HA segments for available avian (n=7) and human (n=13) H7N9 sequences using BEAST (Drummond A J, et al., 2012) (with a strict molecular clock and HKY+Ⅰ substitution model) we found abnormally high substitution rates for the avian samples and broad 95% HPD intervals for both hosts: 0.247 (95% HPD region [1.582E-2, 0.541]) and 2.76E-2 (95% HPD region [9.348E-3, 4.873E-2]) substitutions/site/year for avian and human respectively. Given the limited number of avian samples the high substitution rate for the H7N9 avian samples is not surprising and highlights the need for adequate samples from over a representative time course.
Similarly, the published mutation analyses were limited in that they were primarily comparative insofar as they investigated the published H7N9 sequences for sites known to be associated with virulence or host adaptation (Liu D, et al., 2013; Xiong C, et al., 2013; Zhang W, et al., 2013; Zhu E Z, 2013). However, no new regions or mutations were identified from analysis of the available sequence set.
This highlights not only the importance of surveillance of cases in human and avian populations, but also the need for adequate and balanced sampling to collect information at the genetic level. In the current outbreak, samples were almost exclusively collected from humans and even then relatively sporadically, no new samples were made publicly available once the initial interest had waned (Figure 1). While reporting of human cases provides an overview of how an outbreak is spreading through a population, if this was complemented with sequences from a comprehensive and continuous sampling set from host populations this would allow us to achieve a clearer understanding of how the virus is evolving and adapting to new hosts. In this way, we can better evaluate the potential threat from the emergence of new re-assorted lineages.
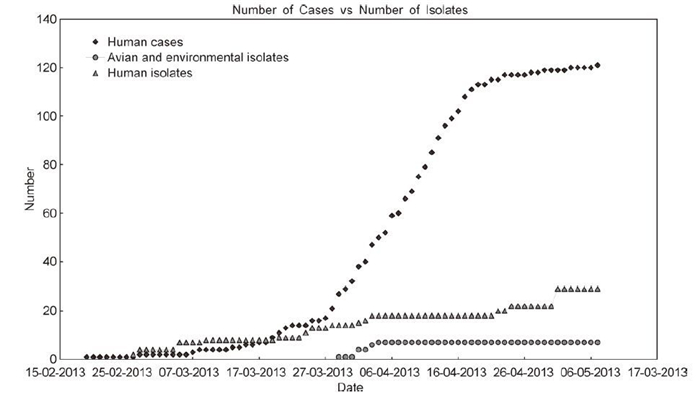
Figure 1. Cumulative number of human cases (upper curve) from H7N9 outbreak over time and corresponding number of published human sequences (middle curve) and avian sequences (lower curve) from collected isolates. Notably, no avian sequences were deposited since the beginning of April.
-
All the authors declare that they have no competing interest. This article does not contain any studies with human or animal subjects performed by the any of the authors.

 DownLoad:
DownLoad: