










HTML
-
Influenza A viruses belong to the Orthomyxovirdae family that cause epidemics in mammalian and bird populations. Over several decades, human infections with avian influenza viruses have raised concerns regarding their pandemic potential worldwide. The reassortant H7N9 and H5N1 viruses contain six internal gene segments that are genetically related to H9N2 virus. Despite the genome similarities, influenza subtypes exhibit differences in their virulence and infectivity. In contrast to H5N1 and H7N9 viruses, H9N2 is low pathogenic, causing mild disease among birds and human (Feng et al., 2013; Guan et al., 1999). Circulation and coexistence of these viruses highlight the need to develop a heterosubtypic vaccine that can provide cross-subtype protection in association with CD8+ cytotoxic T lymphocytes (CTL) and CD4+ responses (Even-Or et al., 2013; Ben-Yedidia and Arnon, 2007; Grebe et al., 2008; Nguyen et al., 2001; Tumpey et al., 2001; Shaw and Palese, 2013). Induction of cross-reactive CTL responses that recognize conserved epitopes of viral antigens shared by different influenza subtypes have been investigated previously by targeting hemagglutinin (HA) or the more conserved nucleoprotein (NP) and matrix protein (M1) (Even-Or et al., 2013; Luo et al., 2012; Qiu et al., 2006; Watanabe et al., 2008). The CTLs recognize multiple viral epitopes presented by major histocompatibility complex (MHC, HLA in human) class Ⅰ on antigen presenting cells (Even-Or et al., 2013; Ben-Yedidia and Arnon, 2007; Belz et al., 2002; Palese and García-Sastre, 2002; Wang, 2010). Among the viral proteins, the highly immunogenic HA, which harbors major neutralizing epitopes, is a promising vaccine candidate (Ben-Yedidia and Arnon, 2007; Even-Or et al., 2013; Bot et al., 1997; Ichihashi et al., 2011; Prabakaran et al., 2010; Zheng et al., 2009). HA peptide vaccine is a potent inducer of CTL, and it confers signifcant protective immunity against influenza infection. Complete heterosubtypic immunity, induced by CD4+ T helper cells, involves B cell-and T-cell-dependent virus-specifc antibody responses mediated by the secretion of cytokines (Nguyen et al., 2001; Tumpey et al., 2001).
A vaccine-formulated adjuvant can greatly modulate T helper responses. Adjuvants employ a variety of mechanisms to enhance immune response. Adjuvants function by prolonging the exposure time of the antigen to the immune system, enhancing the delivery of antigen, directing antigen presentation by the MHC, or providing immunostimulatory signals that potentiate the immune response (Ben-Yedidia and Arnon, 2007; Mastelic et al., 2013). The ability of molecular adjuvants to improve the immunogenicity of vaccines is the focus of intense research (Liniger et al., 2012; Zhang et al., 2000). Secretion of hemokinin-1 (HK-1), a newly discovered member of the tachykinin family, promotes immune cell survival and activation, and induces type-1 CD4+ T cell helper and CD8+ CTL responses (Chen et al., 2012; Nelson et al., 2004; Zhang and Paige, 2003). HK-1 is expressed by human and murine dendritic cells and characterized by the presence of a conserved C-terminal motif, Arg-Ser-Arg-Thr-Arg-Gln-Phe-Tyr-Gly-Leu-Met-NH2. However, the effect of HK-1 on the immune system remains unclear (Zhang et al., 2006). The peptide has been used as a molecular adjuvant to stimulate and enhance humoral and memory responses to hepatitis B surface antigen DNA vaccination (Chen et al., 2012).
One strategy for enhancing influenza epitope-based vaccine protection is to use known epitope targets for eliciting neutralizing antibodies that are capable of neutralizing multiple influenza subtypes (Khanna et al., 2014). However, influenza viruses can undergo extensive genetic and antigenic evolution especially at the HA antigen site (Shahsavandi et al., 2012). Although crystal structures of the viruses have been solved, no details for their antigenic epitopes have been elucidated (Okamatsu et al., 2014). On the other hand, in recent antibody escape mutant examination of H9N2 viruses, novel amino acid positions in the antigenic sites have been revealed, which had not be reported previously (Wan et al., 2014).
In this study, we focused on the HA glycoprotein of avian H9N2, H5N1, and H7N9 subtypes as the inductor of immune response to determine the effect of cross-reactivity on the dynamics of influenza infection in the human population. Because H9N2 virus has high genetic compatibility with H5N1 and H7N9 viruses, we used immunoinformatic tools to analyze the HA protein sequences of the subtypes and selected the predicted conserved T cell epitopes as a means of estimating the immunogenic potential of epitope-based vaccine against the circulating viruses. The heterosubtypic epitope-based vaccine development strategy involved three steps: 1) prediction of conserved high-score CTL epitopes restricted to HLAA*0201-binding peptides within the HA protein of these subtypes, and identifcation off sequences that have the highest potential for HLA-restricted immunogenicity; 2) construction of HA CTL/HK-1 chimera protein models; and 3) in silico modeling analysis of the constructs to identify an ideal epitope-based influenza vaccine candidate.
-
Datasets of HA-deduced amino acid sequences based on circulating H7N9, H5N1, and H9N2 influenza subtypes from their emergence to 2013 were derived from National Center for Biotechnology Information (NCBI) GenBank. Nine-mer sequences corresponding to specifc CTL epitopes restricted to the HLA-A*0201-binding peptides within HA were predicted using the BioInformatics and Molecular Analysis section (BIMAS) HLA Peptide Binding Prediction (http:/bimas.dcrt.nih.gov/molbio/hla_bind/index.html) and SYFPEITHI (http://www.syfpeithi.de/Scripts/MHCServer.dll/EpitopePrediction.htm) websites.
-
Conserved CTL epitopes restricted to the highly prevalent allele HLA-A*0201 allele among the influenza subtypes were selected. To design a single peptide, the amino acid sequences were fused to HK-1 using a repeat of hydrophobic amino acid linkers. The physicochemical properties, hydrophobicity, hydrophilicity, surface accessibility and electrostatic potential of the HA CTL/HK-1 construct proteins were calculated using Prot-Param (http://expasy.org/tools/protparam.html) and CLCprotein Workbench v.5.0.1.
-
To aid alignment correction and loop modeling, the secondary structures of the HA CTL/HK-1 chimera proteins were predicted using PSIPRED (http://bioinf.cs.ucl.ac.uk/psipred). The structures were analyzed using RNAfold (http://rna.tbi.univie.ac.at) to compare their folding and thermodynamic features. Protein structures and three-dimensional (3D) models or tertiary structures of the chimeric constructs were predicted by Phyre (http://www.sbg.bio.ic.ac.uk/phyre2/html/). The 3D models were visualized using Swiss-Pdb Viewer (http://spdbv.vital_it.ch/).
-
The 3D models were constructed based on sequence alignments between the constructs and the template proteins using SWISS-MODEL (Arnold et al., 2006) after energy minimization, computed with GROMOS96. Geometric evaluations of the modeled 3D structures was performed using RAMPAGE (http://mordred.bioc.cam.ac.uk/~rapper/rampage.php) and the structures were validated using ProSA (https://prosa.services.came.sbg.ac.at/prosa.php) (Wiederstein and Sippl, 2007). Homology modeling involves alignment of target sequence, model building, and model quality evaluation using QMEAN4 global score (http://swissmodel.expasy.org) (Benkert et al., 2008). This scoring function describes the major geometrical aspects of protein structures using torsion angles and solvation energies, which play an important role in flexibility (required for folding of the polypeptide backbone) and protein-ligand binding. The possible binding modes between the HK-1 ligand and the HA CTL epitope motifs were computed using the SWISS-MODEL homology-modeling pipeline.
-
The antigenic sites in the chimeric models were predicted using the Immune Epitope Database (IEDB) server (http://tools.immuneepitope.org/tools/bcell/tutorial.jsp). Antigenic epitopes were determined using the antigenicity prediction method described by Kolaskar and Tongaonkar, which is based on the physico-chemical properties of amino acid residues (i.e. hydrophilicity, accessibility, and flexibility) with approximately 75% accuracy. The presence of beta turns in the protein structures was investigated using ProtScale in the Expasy server
-
IFN-γ is the signature cytokine of both the innate and adaptive immune systems, and it elicits antiviral and immune regulatory activities. To design an effective epitope-based vaccine, IFN-γ-inducing MHC class Ⅱ binder peptides from HA CTL/HK-1 were predicted and scanned using the IFN epitope server (http://crdd.osdd.net/raghava/index.php).
HA influenza A virus sequence data collection and epitope prediction
Design and characterization chimeric CTL epitope/HK-1 constructs
Protein structure prediction
Homology modeling and model quality
Antigenic peptide prediction
Interferon (IFN) epitope scanning
-
Specific CTL epitopes restricted to the A*0201 HLA-A supertype were predicted for the HA proteins of H7N9, H5N1, and H9N2 influenza viruses. Three motifs 438VLLENQKTL, 455NLYNKVKRA, and 523KILTIYSTV named A, B, and C, respectively, were conserved between the H5N1 and H9N2 viruses. The HA proteins of H7N9 and H9N2 viruses possessed two similar CTL epitopes, 36TLTENNVPN and 324KLAVGLRNV (D and E, respectively), while a conserved motif was not detected between H7N9 and H5N1. The D and E motifs were attached on both sides of the A/-B/-C peptides using EAAAK repeats, and the resulting peptides were fused to HK-1 (Figure 1). The kozak sequence was introduced to increase the efficiency of translational initiation. Bioinformatics analyses were performed on two HA CTL/HK-1 constructs named Ⅰ and Ⅱ. The physicochemical properties of the constructs are listed in Table 1.
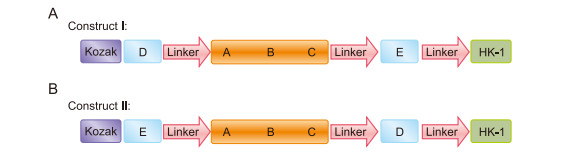
Figure 1. Schematic models of HA CTK/HK-1 constructs (A: construct Ⅰ; B: construct Ⅱ) including the conserved CTL epitopes restricted to the A*0201 HLA-A supertype among the H7N9, H5N1, and H9N2 influenza viruses attached to HK-1.

Table 1. physicochemical properties of HA CTL/HK-1 constructs
-
The mRNA structures of the HA CTL/HK-1 peptides were analyzed using RNAfold, and the minimum free energy levels were estimated to be -32.40 kcal/mol for construct Ⅰ and -31.36 kcal/mol for construct Ⅱ. These values indicate good stability of the mRNA structures. Secondary structure analysis (Figure 2) revealed that construct Ⅱ was composed of random coils (19.72%) and α-helixes (80.28%). Construct Ⅰ had higher α-helixes content (83.10%) and lower random coils content (16.90%) than construct Ⅱ. No β-sheets were detected in these constructs. 3D structures were modeled (Figure 3) and evaluated, and the best model was chosen for further analysis.
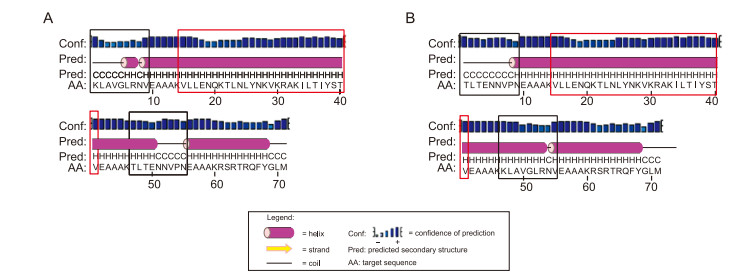
Figure 2. PSIPRED graphical result of secondary structure prediction of the two HA CTL/HK-1 chimera proteins, construct Ⅰ (A) and construct Ⅱ (B). The three conserved CTL epitopes restricted to the A*0201 HLA-A supertype between H5N1, and H9N2 influenza viruses are shown in red and others in black boxes.
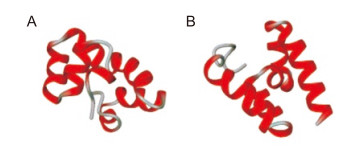
Figure 3. Three-dimensional structure of the HA CTE/HK-1 constructs, construct Ⅰ (A), and construct Ⅱ (B). The images were modeled using the SwissPdb Viewer software.
-
On the basis of the results obtained from the alignment program in SWISSMODEL, the crystal structure of H9 influenza virus (ljsh.1.B) which showed 74.04% sequence identity was selected as the template. To select the best chimeric protein model, the generated 3D HA CTL/HK-1 models were compared. Construct Ⅰ was modeled with 93.3% confdence and 74% identity using the single highest scoring template. Construct Ⅱ was modeled with 94.1% confidence and the same identity as construct Ⅰ. The models were evaluated according to their template modeling scores (TM-scores), root mean square deviations (RMSDs), energy minimization, and Ramachandran plots. The estimated values for both constructs are listed in Table 2. The Ramachandran plots for the two HA CTL/HK-1 peptides were generated to assess the models (Figure 4). The plots revealed that the φ and ψ torsion of angles of all residues in the models were clustered around the secondary structure regions that defne backbone conformation. However, the torsion angles of construct Ⅱ were less widely distributed than those of construct Ⅰ, and they showed better clustering within the allowed regions of the plot.

Table 2. The QMEAN4 Z-score, TM-score and homology modeling RMSD of HA CTL/HK-1 chimeric proteins
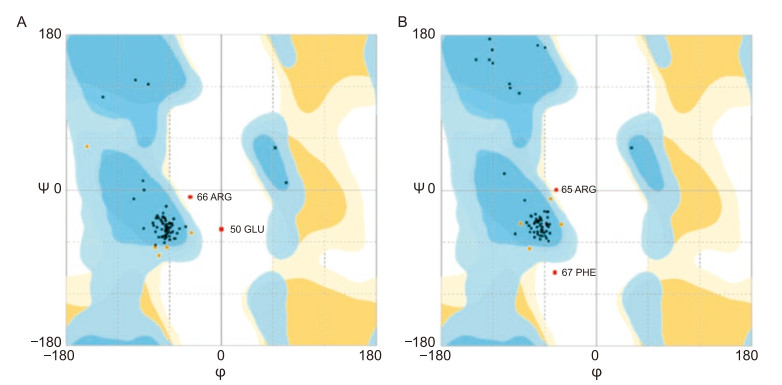
Figure 4. Evaluation of HA CTL/HK-1 model quality by Ramachandran plot analysis. (A) construct Ⅰ: 85.1% of the residues were present in favored region, 10.8% in the allowed region, and 4.1% in the outlier region; (B) construct Ⅱ: 91.9% of the residues were in favored region, 2.7% in the allowed region, and 5.4% in outlier region. The φ and ψ torsion angles of amino acid residues in the proteins were reasonably accurate.
The overall quality of the models was validated; Z-scores of -3.63 and -4.27 were obtained for constructs Ⅰ and Ⅱ, respectively (Figure 5). Local geometry was evaluated on the basis of torsion angle potential, atomic distance-dependent interaction potential, and buried status of the residues (solvation free energy), using the QMEAN Z-score (Table 2). The higher Z-score (-1.65) obtained for construct Ⅰ may be attributed to differences in the torsion and solvation energies of the two constructs.
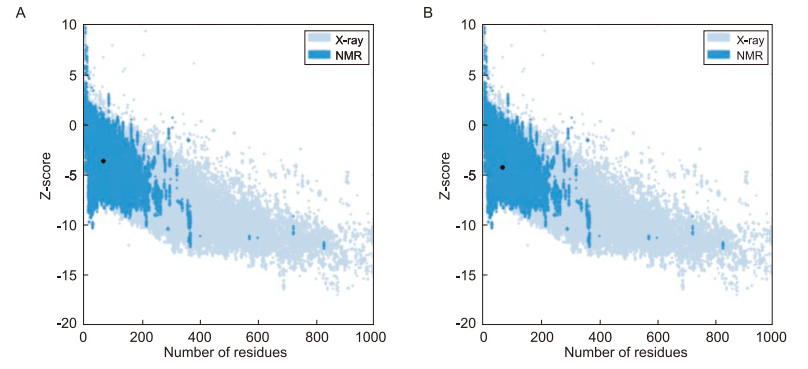
Figure 5. ProSA model quality of the two HA CTL/HK-1 chimera proteins, construct Ⅰ (A, Z-score –3.98) and construct Ⅱ (B, Z-score –4.73).
-
According to the Kolaskar and Tangaonkar antigenicity scale, the most likely antigenic determinants for construct Ⅰ were at positions 12-18, 23-29, and 31-58, with an average antigenic propensity of 1.028. For construct Ⅱ, the antigenicity plot predicted antigenic determinants at regions 4-18, 23-29, and 31-46 with an average antigenic propensity of 1.021 (Figure 6). According to the Levitt scale, amino acid residues at these positions fall inside the beta turn region (1.000 as the threshold level). The predicted segments from both sequences may be antigenic, and may elicit antibody response.
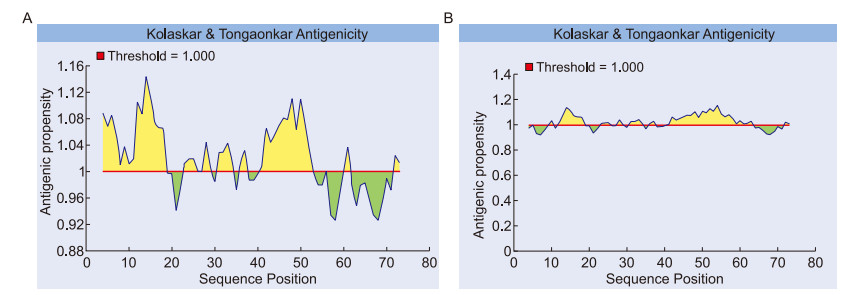
Figure 6. Antigenicity prediction plot of HA CTL/HK-1 chimera peptides, predicted using the Kolaskar-Tongaonkar method. Regions with antigenic propensity scales > 1 are predicted as antigenic regions. The average antigenicity of construct Ⅰ (A) was 1.028 at the 12AAKVLLE18, 23LEAAAKN29, and 31AKKILTIYSTVKLAVGLRNVEAA58 motifs. The average antigenicity of construct Ⅱ (B) was 1.021 at the 4VGLRNVEAAAKVLLE18, 23LEAAAKN29, and 31AKKILTIYSTVT46 motifs.
-
All possible mutant chimeric peptides were generated by a mutating single residue at a time, and the ability of each mutant to induce IFN-γ was predicted. Sorting of the mutants revealed that replacement of T at position 1 with K increased the probability of inducing IFN-γ from -4.352 to 2.572 in construct Ⅰ. Substitution of E at position 50 with F, H, K, L, M, Q, R, W, or Y in construct Ⅱ raised the score from -4.352 to 3.0. The HLA-A*0201-binding properties of the altered epitopes were predicted, and the results revealed that the substitutions had no effect on the HLA-binding affnity.
Construction of chimeric HA CTL/HK-1
Protein structure prediction
Homology modeling and model quality evaluation
Antigenicity evaluation
IFN-γ scanning of the chimeric HA CTL/HK-1 IFN models
-
The coexistence of different influenza subtypes in the same susceptible population is likely to generate conditions for the emergence of novel reassortant strains leading to limited protection against viral infection. Different approaches, including subtractive genomics and predicting HLA-binding motifs, have been adopted to identify conserved epitopes within influenza proteins. Identifcation of potential epitopes within antigenic influenza proteins might open opportunities for the development a peptide vaccine that elicits heterosubtypic T-cell responses (Baker and Sali, 2001; Cheung et al., 2010; Danke and Kwok, 2003; Grebe et al., 2008; Palese and García-Sastre, 2002; Staneková and Vareckova, 2010; Sun et al., 2010). The binding affnity of the HLA haplotype to individual epitopes is the main factor responsible for the immunodominance of CTL epitopes. The most frequently recognized conserved epitopes of influenza antigens restricted by HLA-A*0201 occur at high frequencies in Northern and Southeast Asia and North American populations (Baker and Sali, 2001; Cheung et al., 2010).
Here, we describe a heterosubtypic vaccine model based on the conserved CTL epitopes of the HA proteins of the H5N1, H9N2, and H7N9 influenza viruses. Epitope distribution analysis revealed that the two conserved HA CTL epitopes between H7N9 and H9N2 viruses occur in the HA1 and HA2 subunits, whereas the three conserved epitopes between H5N1 and H9N2 occur in the HA2 subunit. Two peptide constructs were designed based on the shared CTL epitopes among the subtypes. We used the immune regulation peptide, HK-1, to improve immune responses against influenza viruses. In silico analysis was performed to examine the applicability of the CTL epitope-HK1 in developing a vaccine capable of stimulating effective T-cell response. As shown in Table 1 the physicochemical properties of the two constructs were similar. The instability index categorized both proteins as stable. As shown in Figure 2, secondary structure analysis revealed lower random coil and higher α-helical contents in construct Ⅰ than that in construct Ⅱ. The regions with a high flexibility are potential epitope regions and increased epitope flexibility may enhance the binding ability of antibody. Although, random coils play an important role in the high flexibility of proteins, it is important to note that the most functional elements in HA of influenza viruses are contained within α-helix and the existence of the structure in the N-and C-terminals of a protein shows that the protein had good flexibility, too. The additional α-helixes may play an important role in DNA binding and elicit stability.
To determine the possible function of HA/CTL, the sequences were subjected to comparative protein structure modeling using the target protein sequence. SWISSMODEL was used to build the models and for global energy minimization. The topological similarity of two peptide structures was assessed by calculating their TMscores. RMSD was calculated to measure the average distance between the backbones of the superimposed proteins. The RMSD values of the modeled constructs were different (Table 2). Values greater than 0.15 are considered to represent significant structural perturbations, that might have functional implications for the protein. In a review, Baker and Sali (2001) described the usefulness of RMSD quality estimation for predicted models. They suggested that a model with an RMSD of 1 to 1.5Å is appropriate for structure-based protein engineering applications such as drug design. However, a model with RMSD of approximately 4Å, where residue positions are mostly correct, is still useful for drug design and for performing small ligand docking predictions. For protein topology classifcation, a TM-score of approximately 0.5 is considered the threshold value to decide whether two structures are of the same fold or not (0.0 < TM-score < 0.17 indicates random structural similarity and 0.5 < TM-score < 1.00 indicates same fold). In our study, a length-independent TM-score of ~0.4 for construct Ⅱ was the best possible converge that could be obtained. This may be attributed to the high sequence similarity between the alignment models. Because the folds of both models were expected to be correct, the functions of the proteins were analyzed using different programs, including PROCHECK. Ramachandran plot quality was evaluated, and the QMEAN4 scores were calculated. The Ramchandran plot for construct Ⅱ revealed that 91.1% of the residues were present in the most favored regions, 2.7% in the allowed regions, and 5.4% in the disallowed regions. In the case of construct Ⅰ, the distribution was 85.1%, 10.8%, and 4.1%, respectively. The Ramachandran plot of φ and ψ torsion angle distribution in the models was computedusing PROCHECK to checking for non-glycine residues in the disallowed regions. The angles were reasonably accurate, including geometrically acceptable quality for both models.
ProSA-web was used to validate the models. This program compares and analyzes energy distribution in a protein structure as a function of sequence position. As shown in Figure 5, the Z-scores thus obtained suggested that, these models were of good qualityn. QMEAN4 scoring (Benkert et al., 2008) was used to analyze local geometry by calculating torsion angle, distance-dependent interaction potentials based on C-beta and all-atom levels, and solvation energy. These parameters were different in both constructs, and construct Ⅰ showed better scores for torsion angle and solvation energy. Since the overall quality Z-score of construct Ⅰ improved to -1.65, this chimeric peptide was chosen as the fnal model, and its ability to induce heterosubtypic immune responses against influenza viruses was evaluated. To confirm the ability of HA/CTL to elicit immune response, the antigenic nature of the peptide was determined using the Kolaskar and Tongaonkar method. The predicted fragments occurred within β-turns, which play an important role in protein folding and stability. Moreover, these fragments probably interact with MHC molecules. The IFN-γ inducing potential of the chimeric peptide increased when lysine was substituted at defned positions. These substitutions had no effect on the HLA-A*0201 binding affnity of the epitope.
Previous studies have shown that the HA protein is a promising candidate for the design of a vaccine construct aimed at providing broad-spectrum immunity to influenza viruses. However, many researchers have focused on protective T cell responses to NP and M2 antigens because of frequent point mutations in HA. NP is a major target of immunodominant CD8+ T-cell responses (Grant et al., 2013), but immunizations with internal proteins confer different levels of protection against lethal challenge in mice, and they likely dependent on the mode of gene-based vaccination, dose of challenge virus, and formulation with adjuvant (Jimenez et al., 2007; Epstein et al., 2005). Recently, Rao et al. (2013) investigated the effcacy of highly conserved proteins compared to HA in protecting against H5N1 virus in ferrets, and they suggested that these antigens require combinatorial vaccination with HA to induce protection against the strain. The cross-protective potential of these viral peptides could be increased by optimization of their delivery and immunogenicity using adjuvants. In this study, we designed epitope-based vaccine models by incorporating fve conserved HA CTL motifs from three circulating influenza subtypes. We predicted the biological effects of HK-1 on immunological regulation. Moreover, we incorporated a molecular adjuvant into the peptide constructs to enhance the immunogenicity of the vaccine. The ability of the chimeric proteins to induce immune responses was evaluated using different bioinformatics approaches, and fnally, one construct was validated as a suitable model. The heterosubtypic protective immunity of the HA/CTLHK-1 proteins against influenza viruses and the immunostimulatory effect of HK-1 on the chimeric HA protein needs to be investigated in future studies.
-
This work was supported by Razi Vaccine & Serum Research Institute, Karaj, Iran.
-
The authors declare that they have no competing interest. This article does not contain any studies with human or animals subjects performed by any of the authors.
-
S Shahsavandi conceived the study, analyzed the data and wrote the manuscript. MM Ebrahimi, K Sadeghi and M Mahravani participated in data analysis.

 DownLoad:
DownLoad: