









HTML
-
Owing to their high lytic activity against multi-drug resistant (MDR)bacterial pathogens, bacteriophage lysins are promising candidates for application as enzybiotics (Ahluwalia and Sekhon, 2012; Fischetti, 2010; Pastagia et al., 2013). These peptidoglycan hydrolases expressed by phages in the late phase of the infection cycle have been identified as effective anti-infectious agents in various animal models(Junjappa et al., 2013; O'Flaherty et al., 2009; Singh et al., 2014; Veiga-Crespo et al., 2007; Yang et al., 2014a). Recent research has shown that engineered "Artilysins" can lyse Gram-negative pathogens directly from without(Briers et al., 2014; Lukacik et al., 2012), thus extending the application field of lysins. Lysins usually display a typically modular structure with at least two distinct domains(Vill and Crespo, 2010), specifically, an N-terminal catalytic domain(CD)responsible for enzymatic hydrolysis and a C-terminal cell wall binding domain(CBD)conferring substrate recognition specificity. However, the genus specificity of lysin may be a barrier for application in mucous-associated infections, since human mucous membranes are a reservoir for several pathogenic ba cteria, including pneumococci, staphylococci and streptococci (Coello et al., 1994; De Lencastre et al., 1999; Fischetti, 2003). Therefore, lysins with a broad s pectrum of lytic activity would be of considerable therapeutic value.
A limited number of lysins with broad lytic spectra have been identified to date, such as PlyV12(Yoong et al., 2004) and PlySs2(Gilmer et al., 2013). Development of chimeric lysins(chimeolysins)through domain shuffling may effectively extend the lyti c spectrum(Yang et al., 2014a; Yang et al., 2014b). For instance, a previous study by our group showed that the CBD of PlyV12 has broad binding capacity, and the lytic spectrum of Ply187N could be extended through fusing with the CBD from PlyV12(Dong et al., 2014), indicating that CBDs with binding capacities to multiple genera are good donors for lysin engineering. To our knowledge, only CBD of PlyV12 has been confirmed with broad binding capacity so far.
PlySs2, derived from a Streptococcus suis phage, is active against multiple genera, including staphylococci, streptococci(GAS, GBS, GGS, GES) and Listeria (Gilmer et al., 2013; Schuch et al., 2014). In the present study, lytic activity of the catalytic domain(CD, PlySc) and binding specificity of the cell wall binding domain (CBD, PlySb)of PlySs2 were assessed, with a view to establishing the determinants of its broad lytic spectrum.
-
All strains used in this work are described in Table 1. Streptococcus suis, S. dysgalactiae, S. pyogenes, Listeria and Enterococcus faecium strains were cultivated in brain heart infusion(BHI)broth, and the remaining strains in Luria-Bertani(LB)broth.

Table 1. Strains used in this study
-
The original plySs2 gene was chemically synthesized into pUC57 plasmid by Sangon Biotech(Shanghai, China). To assess the binding specificity of PlySs2, seven truncated fractions were constructed and genetically fused with an N-terminal enhanced green fluorescent protein(EGFP)cloned from pET-EGFP, generating the recombinant proteins EGFP-PlySs2, EGFP-PlySc, EGFP-PlySb, EGFP-62PlySb, EGFP-41PlySb, EGFP-35PlySb and EGFP-27PlySb, respectively. PlySs2 and its putative CD(PlySc)were additionally cloned and purified for examination of lytic activity. The primers used in this study are shown in Table 2. All gene fragments were cloned into pET28a(+)vector and transformed into E. coli BL21(DE3)cells for protein expression after verification via sequencing.

Table 2. Primers used in this study
-
Recombinant proteins were purified using His tag as described previously(Yang et al., 2012), with minor modifications. In brief, cells were cultured in LB containing 50 μg/mL kanamycin to OD600 of 0.4–0.6. After cooling to 16 ℃, expression was induced for 12 h via addition of isopropyl-β-D-thiogalactopyranoside(IPTG) to a final concentration of 0.2 mmol/ L. Cells were harvested and resuspended in buffer A(20 mmol/L Tris-HCl, 500 mmol/L NaCl, 20 mmol / L imidazole, pH 8.0) with 30% glyc erol. After soni cation, the supe rnatant was collected by centrifugation at 12, 000 g for 30 min at 4 ℃. Purification was achieved following the general protocol using a nickel nitrilotriacetic acid column, and subsequent washing and elution with imidazole solutions at concentrations of 40 and 400 mmol/ L, respectively. Collected fractions were dialyzed against buffer B(20 mmol/L Tris-HCl, pH 7.4)with 30% glycerol and then stored at -80 ℃ until use after quantitation using sodium dodecyl sulfate-polyacrylamide gel electrophoresis (SDS-PAGE) and Bradford assay.
-
Lytic activities of PlySc, PlySs2, EGFP-PlySc and EGFP-PlySs2 were confirmed using the turbidity reduction assay described previously(Gilmer et al., 2013), with minor modifications. Cells were washed once and resuspended in buffer B to a final OD600 of 1.0. Aliquots of 100 μL suspensions were mixed with PlySc, PlySs2, EGFP-PlySc or EGFP-PlySs2(each at a final concentration of 926 pmol/mL)in a 96-well plate, and changes in OD600 monitored immediately using a microplate reader (Synergy H1, BioTek, USA)at 37 ℃ for 60 min. Wells containing mixtures of bacterial cells and buffer B were used as controls. Lytic activity was calculated as initial OD600 divided by final OD600 in each well. All experiments were performed for three replicates.
To determine the binding activities of the EGFP-tagged fractions, mid-log phase cells were washed twice with phosphate-buffered saline(PBS, 137 mmol/L N aCl, 2.7 mmol/L KCl, 4.3 mmol/L Na2HPO4 · H2O, 1.4 mmol/L KH2PO4, pH 7.4) and mixed with 10 mmol/L EG FP, EGFP-PlySc, EGFP-PlySb, EGFP-62PlySb, EGFP-41PlySb, EGFP-35PlySb or EGFP-27Pl ySb at 37 ℃ for 30 min. Staining of EGFP-PlySs2 was performed in phosphate buffer with 750 mmol/L NaCl(PB, 750 mmol/L NaCl, 2.7 mmol/L KCl, 4.3 mmol/L Na2HPO4 · H2O, 1.4 mmol/L KH2PO4, pH 7.4)to suppress lytic activity. Subsequently, cells were washed twice with PBST buffer (PBS with 0.5% Tween-20) and resuspended in PBS, prior to fluorescence microscopy analysis(Delta Vision Personal DV, Applied Precision, USA).
Bacterial strains
Construction of expression vectors
Expression and purification of recombinant proteins
Activ ity assays
-
The full-length PlySs2 protein consists of 245 residues that form an N-terminal CHAP domain(1–146 aa) and a C-terminal SH3-5 cell wall binding domain(162–218 aa), as observed from BLAST analysis. To clarify the mechanism underlying the broad lytic spectrum of PlySs2, the CHAP(PlySc) and CBD(PlySb)domains of PlySs2 were separately constructed and purified(Figure 1). Intact PlySs2, PlySc and several truncated fragments of PlySb were additionally fused with EGFP, and the binding activities evaluated. Recombinant proteins were efficiently expressed in E. coli and used for lytic activity and binding specificity assays following dialysis.
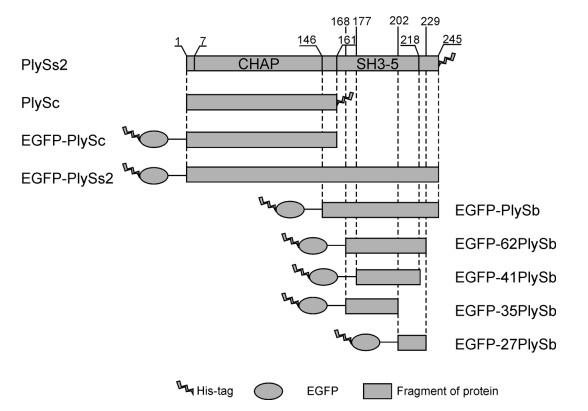
Figure 1. Schematic representation of the recombinant proteins expressed in the study.
-
Purified PlySs2 displayed high lytic activity against various bacteria, including staphylococci and streptococci, but not the Listeria strain examined. Fusion to EGFP had a minor influence on the lytic activity of recombinant PlySs2(Figure 2A). However, the catalytic domain alone, PlySc, showed significantly reduced lytic activity against S. suis(Figure 2B), and fusion with EGFP (EGFP-PlySc)almost abolished lytic activity(data not shown). The weak activity and reduced lytic spectrum of PlySc clearly indicate that the function of PlySs2 is CBD-dependent.
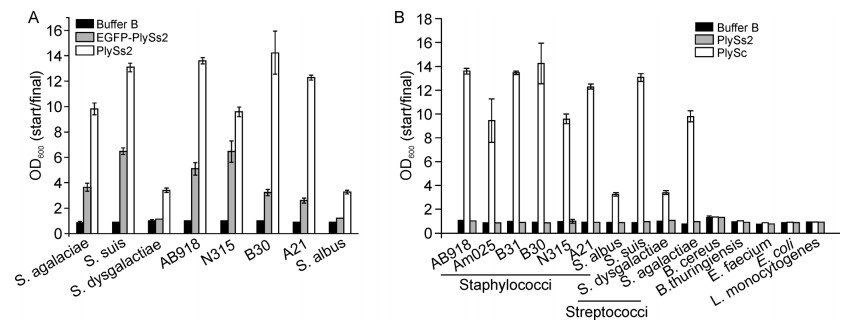
Figure 2. Lytic activity of EGFP-PlySs2, PlySs2 and PlySc. Cells were washed once and treated with 926 pmol/mL protein. (A) Lytic activity of EGFP-PlySs2 and PlySs2 against strains resuspended in buffer B. (B) Lytic activity of PlySs2 and PlySc against various strains.
-
Initially, we examined the binding specificities of EGFP-PlySs2 and EGFP-PlySc. To avoid the potential lytic activity of EGFP-PlySs2 on the strains examined, staining was performed in phosphate buffer containing 750 mmol/L NaCl(PB). As shown in Figure 3, the lytic activity of EGFP-PlySs2 was markedly suppressed in PB containing 750 mmol/L NaCl. Fluorescence microscopy revealed that EGFP-PlySs2 displays high affinity for streptococci, but not S. aureus strains(Figure 4), while binding activity to all strains was abolished in EGFPPlySc (data not shown).
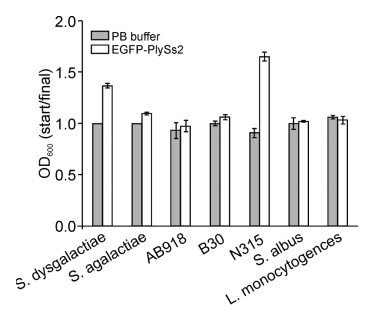
Figure 3. Lytic activity of EGFP-PlySs2. Lytic activity of EGFP-PlySs2 against strains resuspended in phosphate buffer containing 750 mmol/L NaCl (PB).
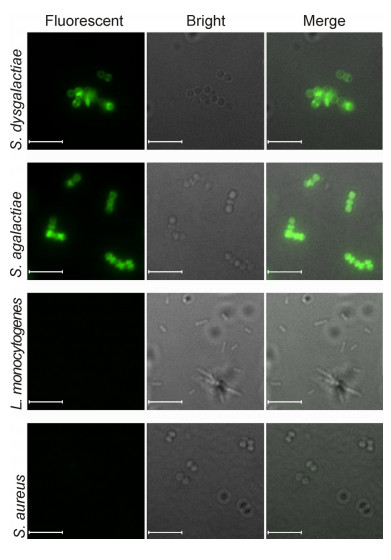
Figure 4. Binding characteristics of EGFP-PlySs2. Cells were mixed with 10 mmol/L EGFP-PlySs2 in phosphate buffer containing 750 mmol/L NaCl (PB) for 30 min at 37 ℃ and analyzed via fluorescence microscopy after washing twice with PBST. All images were obtained under the same instrument conditions. Bar size, 5 μm.
Next, the binding capacity of EGFP-PlySb was ascertained via examination of the fluorescence of lysed bacteria. The results showed that EGFP-PlySb recognizes the streptococcal strains tested, including S. suis, S. agalactiae and S. dysgalactiae, but not S. aureus(Figure 5). To further validate that binding of EGFP-PlySb to staphylococci is abolished, five other clinical S. aureus strains were stained with EGFP-PlySb. As expected, no fluorescence was observed(data not shown).
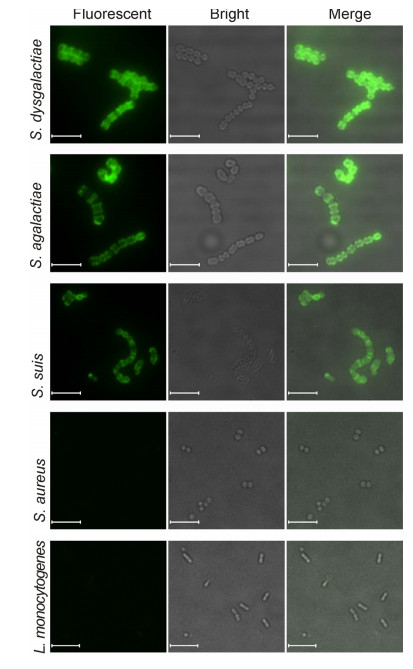
Figure 5. Binding characteristics of EGFP-PlySb. Cells were mixed with 10 mmol/L EGFP-PlySb at 37 ℃ for 30 min and analyzed via fluorescence microscopy after washing twice with PBST. All images were obtained under the same instrument conditions. Bar size, 5 μm.
To determine the core sequence required for the specific recognition of PlySb, four truncated fragments(EGFP-62PlySb, EGFP-41PlySb, EGFP-35PlySb and EGFP-27PlySb)were constructed, and their binding activities for S. dysgalactiae tested. All truncated fragments, alone or mixed, showed no binding activity. Our data collectively indicate that intact PlySs2 has a genus-specific binding capacity for streptococci, but not staphylococci. Moreover, the integrity of PlySb is necessary for the binding capacity of PlySs2.
Expression and purification of recombinant proteins
Lytic activity assay
Binding specificity assay
-
PlySs2 is the first unique lysin reported to kill streptococci, staphylococci as well as Listeria. In the current study, we obtained new insights into the factors contributing to the broad lytic activity of PlySs2 by characterizing the CD and CBD domains of PlySs2. As shown in Figure 3, the CHAP CD domain(PlySc)alone displayed not only a narrow lytic spectrum but also significantly weaker lytic activity, compared with PlySs2. In the binding assay, only fragments containing the full-length SH3-5 CBD domain of PlySs2(EGFP-PlySs2 and EGFPPlySb) showed obvious binding fluorescence to streptococ ci(S. suis, S. agalactiae and S. dysgalactiae), but not staphylococci or the Listeria strains tested(Figures 4 and 5). In view of the high staphylolytic and streptolytic activities of PlySs2(Figure 2), the results suggest that both CD and CBD domains are essential, not only for lytic activity, but also the broad lytic spectrum of PlySs2.
Notably, while PlySs2 displayed high lytic activity against staphylococci, binding was abolished in EGFPPlySs2 and EGFP-PlySb, and lytic activity in PlySc. This finding is inconsistent with data obtained with other lysins so far. Some lysins are reported to contain a non-active CD and active CBD upon sole expression of these domains, similar to the streptococcal lysin PlyC (McGowan et al., 2012; Nelson et al., 2006). For other lysins, such as Ply187, the CD(Pc)alone has been shown to display higher lytic activity against S. aureus than full-length lysin(Loessner et al., 1999). The lytic activity of Pc could be further enhanced by adding other CBDs(Mao et al., 2013; Yang et al., 2014b). Further research on the interactions between PlySb and PlySc is warranted to elucidate the mechanisms underlying the lytic activity of PlySs2 against staphylococci.
Another interesting characteristic of PlySs2 is its high lytic activity against two L. monocytogenes(HER 1083 and HER 1184)out of the seven strains tested(Gilmer et al., 2013), which is not observed with other lysins from non-Listeria phages. In the current study, neither PlySs2 nor PlySc could lyse the L. monocytogenes strain tested. Moreover, no binding fluorescence to the strain was observed for EGFP-PlySs2 or EGFP-PlySb, further confirming that PlySs2 acts on limited L. monocytogenes strains. One possible reason for the selective lack of activity of PlySs2 on the L. monocytogenes strain examined here may due to its different modifications in the bacterial cell wall, compared with those of the HER1083 strain. The receptor for PlySs2 in the bacterial cell wall remains to be identified.
As an initial step in testing the compatibility of PlySb as a CBD donor, we replaced the CD of PlySs2 with that of Ply118, Ply511(Listeria bacteriophage lysins), and LysH5(staphylococcal phage lysin). The lytic spectra of these chimeras were restricted to their parental CD, but not extended to streptococci(data not shown). Therefore, PlySc appears essential for lytic activity on streptococci, the cell walls of which cannot be lysed by other CDs. In conclusion, PlySb and PlySc act synergistically to kill susceptible bacteria. Results from the current study should aid in engineering of PlySs2 for the development of more effective lysins.
-
This work was supported by the National Natural Science Foundation of China(No. 31400126), Basic Research Program of the Ministry of Science and Technology of China(No. 2012CB721102), Chinese Academy of Sciences(No. KJZD-EW-L02), Open Research Fund Program of the State Key Laboratory of Virology of China(No. 2014IOV002), and Key Laboratory of Emerging Infectious Diseases and Biosafety, Wuhan.
-
All the authors declare that they have no conflict of interest. This article does not contain any studies with human or animal subjects performed by any of the authors.
-
HP Wei and H Yang conceived the study and analyzed the data. YL Huang and H Yang performed the experiments. JP Yu participated in data analysis. YL Huang, H Yang and HP Wei wrote the manuscript.

 DownLoad:
DownLoad: