









HTML
-
The original description of a unique skin lesion by Moritz Kaposi in 1872 predates the foundation of viral oncology (Kaposi, 1872). Over 100 years later, an epidemic of this disease, Kaposi's sarcoma(KS), was the first indicator of the devastating HIV/AIDS epidemic to follow (CDC, 1981; Gottlieb et al., 1981). The discovery of the causative agent of KS, Kaposi's sarcoma-associated herpesvirus(KSHV), is one of the great successes of modern biomedical research (Chang et al., 1994; Ganem, 2010; Moore and Chang, 2010; Chang and Moore, 2014). KSHV is also etiologically linked to two lymphoproliferative disorders, primary effusion lymphoma(PEL) and multicentric Castleman's disease(MCD) (Soulier et al., 1995; Cesarman et al., 1996). In the past 20 years, molecular, clinical, and epidemiological studies have generated vast amounts of data, and facilitated our current understanding of KSHV and its associated diseases. A comprehensive description of the pathobiology and epidemiology of KS is beyond the scope of this review, and has been elegantly summarized by several review articles (Ganem, 1997; Verma and Robertson, 2003; Ganem, 2006; Cesarman and Mesri, 2007; Dittmer and Damania, 2007; Ganem, 2007; Ganem, 2010; Mesri et al., 2010; Uldrick and Whitby, 2011; Dittmer and Damania, 2013; Gramolelli and Schulz, 2015). Instead, we will briefly introduce the biology of KSHV pertinent to the following sections on the KSHV epigenome and model systems to study KSHV.
KSHV is a large(~165 kb), double-stranded DNA(dsDNA) virus of the subfamily gammaherpesvirinae (Figure 1). Its closest relatives, herpesvirus saimiri(HVS), Rhesus monkey rhadinovirus(RRV), and murid herpesvirus 68(MHV-68), are important model systems for the study of gammaherpesviruses. The closely related gammaherpesvirus, Epstein-Barr virus(EBV), is also intensively studied, since it is a ubiquitous and potentially oncogenic human pathogen (Young and Murray, 2003; Raab-Traub, 2012). Herpesviral particles are composed of a linear dsDNA genome enclosed by an icosahedral protein capsid, which is enveloped by a glycoprotein-studded lipid bilayer. An intermediate layer between the capsid and envelope is referred to as the tegument. KSHV can infect a variety of cell types, and enters cells either through fusion or receptor-mediated endocytosis (Chandran, 2010; Chakraborty et al., 2012; Veettil et al., 2014). Once the genome is delivered into the host nucleus, KSHV, like all herpesviruses, is capable of entering into one of two alternative life cycles: latency or lytic replication.

Figure 1. The KSHV episome. Kaposi's sarcoma-associated herpesvirus (KSHV) encodes 87 open reading frames (ORFs) and at least 17 microRNAs (purple boxes), 14 of which are co-expressed as a cluster. A striking feature of KSHV is the number of ORFs (at least 14) that encode cellular orthologues (yellow boxes). Putative latent transcripts are indicated in green. KSHV exists as an episome (double-stranded circular DNA) within the host nucleus. Reactivation can occur when the promoter of ORF50 is activated, resulting in the expression of replication and transcription activator (RTA), the main regulator for the viral lytic replication programme. Early lytic genes include those encoding viral proteins required for DNA replication or viral gene expression, whereas late lytic genes are those encoding viral structural proteins, such as envelope and capsid proteins, that are required for assembly of viral particles (virions). Polyadenylated nuclear (PAN) RNA is transcribed from a locus residing between K6 and ORF16. The terminal repeat (TR) sequences account for ~20–30 kb. Adapted by permission from Macmillan Publishers Ltd: Nature Reviews Cancer (Mesri et al., 2010) © 2010.
In the context of oncogenesis, both serve critical roles. Latency, during which only a few key genes are substantially expressed, is required for maintenance of the viral genome(episome) within cells and thus, persistent infection. Lytic replication, in which the entire complement of genes is expressed in a temporally regulated cascade leading to production of progeny virions, is also crucial, because (ⅰ) lytically infected cells serve as a reservoir of new infectious viral particles; and (ⅱ) paracrine signals induced by lytic gene products contribute to inflammation and angiogenesis (Ganem, 2010). Elucidating the factors that influence the gene expression program employed by KSHV upon either primary infection or the latent-lytic switch are of major interest to KSHV researchers and paramount relevance to KSHV pathobiology. Accumulating evidence suggests that epigenetic regulation plays a crucial role in dictating the outcome of KSHV infection.
-
In the past decade, our understanding of epigenetic phenomena has expanded tremendously. This is due, in large part, to the development and application of tools and techniques to study patterns of DNA methylation, protein-DNA interactions, post-translational modifications, gene expression, and higher-order chromatin structure, as well as unprecedented advances in sequencing technology. With regard to KSHV, the wealth of information that has emerged provides insights into the roles of chromatin organization throughout the viral life cycle. Together, these data indicate that epigenetic regulation is intimately linked to the fate of KSHV and its human host.
-
Herpesviral genomes are thought to exist in one of three general states of primary chromatin structure:(ⅰ) as naked DNA within capsids; (ⅱ) regularly chromatinized(packaged with cellular histone proteins to form nucleosomes, the fundamental repeating units of chromatin) during latency; or (ⅲ) in an intermediate 'lytic state' characterized by dynamic alterations in chromatin architecture (Paulus et al., 2010; Nevels et al., 2011). Upon infecting cells, herpesviruses must transport their genome to the nucleus, where it is circularized and chromatinized, resulting in the formation of stable, non-integrated episomes with similar structure to the host genome (Lieberman, 2013). Genome circularization is critical for viral episome maintenance, DNA replication, and evasion of the host DNA damage response (Ballestas et al., 1999; Strang and Stow, 2005; Deng et al., 2012). Chromatinization after primary infection is also a major determinant of the fate of KSHV. This is intuitive, since the precise nature of nucleosome positions and epigenetic modifications potentiates replication, recombination, repair, and transcription of the underlying DNA (Bannister and Kouzarides, 2011). Early findings that KSHV stable episome maintenance is not very efficient indicated a role of epigenetic factors in persistent infection (Grundhoff and Ganem, 2004). It is likely that inefficient circularization or chromatinization accounts for the high failure rate in the establishment of stable infections by gammaherpesviruses(KSHV and EBV) (Hurley and Thorley-Lawson, 1988; Darst et al., 2013).
Several recent studies have characterized the epigenetic landscape of KSHV, both during latency and lytic replication. This has led to the identification and characterization of many viral and host factors responsible for modulating the viral epigenome. The high-resolution study of epigenomic marks, including DNA methylation, histone post-translational modifications, nucleosome position, and 3D chromatin organization, has been facilitated by the development of powerful sequencing-based methods, including Chromatin Immunoprecipitation(ChIP), a versatile and efficient technique used to investigate protein-DNA interactions. ChIP has been combined with DNA microarrays(ChIP-on-Chip) or sequencing(ChIP-seq) to offer high-throughput, genome-wide characterization of the extent of histone modifications and/or histone-modifying enzymes at specific genomic loci, or with chromatin conformation capture(3C) assays to investigate higher-order chromatin structure. Recently, next-generation sequencing following Formaldehyde-Assisted Isolation of Regulatory Elements(FAIRE-seq) has also served as a viable alternative to ChIP-seq assays. The following sections will discuss the novel and salient findings regarding epigenetic regulation in the context of KSHV replication.
-
DNA methylation, typically occurring at CpG sites in mammalian cells, are associated with gene silencing (Jin et al., 2011; Jones, 2012). Unsurprisingly, the level increases over time following KSHV de novo infection, presumably contributing to repressed viral gene expression (Gunther et al., 2014). A study coupling immunoprecipitation of methylated DNA(MeDIP) to a KSHV tiling microarray characterized the DNA methylation pattern of the KSHV genome in latently-infected cells (Gunther and Grundhoff, 2010). In general, the data suggested an inverse correlation between DNA methylation and KSHV gene expression(i.e., reduced DNA methylation at transcriptionally active sites, such as the latency locus, while most lytic gene loci were methylated). Interestingly, DNA methylation appears to play a role in the control of latency, since a DNA methyltransferase inhibitor, 5-azacytidine(5-AzaC) triggers lytic reactivation (Pantry and Medveczky, 2009). Consistent with this role, a KSHV-encoded microRNA, miRNAK12-5, has been shown to increase global levels of viral/cellular DNA methylation, thereby preventing lytic gene expression (Lu et al., 2010). A more recent study employing MAPit(Methylation Accessibility Probing for individual templates) single-molecule footprinting found that the status of DNA methylation is incredibly diverse at specific loci across a population of KSHV episomal genomes (Darst et al., 2013).
-
Nucleosome position and post-translational modifications of histone tails are widely recognized predictors of gene expression, since they affect the local chromatin architecture and accessibility of transcription factors (Kornberg and Lorch, 1999; Karlic et al., 2010; Tsankov et al., 2010). The putative effect(s) on transcriptional regulation depends on the histone residue modified, and the specific modification(i.e., methylation, acetylation, phosphorylation, ubiquitylation, sumoylation, etc.). The first clues that histone modifications possess functional roles in the KSHV life cycle came from the findings that the histone deacetylase(HDAC) inhibitor sodium butyrate(NaB) and the histone acetyltransferase(HAT) inducer, tetradecanoylphorbol acetate(TPA) potently stimulate KSHV lytic reactivation (Renne et al., 1996; Wang et al., 2003; Miller et al., 2007). In 2010, two independent studies used ChIP-on-Chip to reveal the epigenetic landscape of KSHV episomes in infected cells. The first of these analyzed DNA methylation(discussed above) and histone modifications in PEL-and KS-derived cell lines during latency (Gunther and Grundhoff, 2010). Interestingly, the widespread distribution across latent genomes of the bivalent mark, H3K27me3, which is capable of transcriptional repression despite the presence of activating marks(i.e., H3K9/K14ac and H3K4me3), suggested a poised state of repression, allowing for rapid and robust induction of transcription upon lytic reactivation. Indeed, Toth et al. analyzed the same activating and repressive histone modifications during latency and lytic reactivation, and observed this bivalent chromatin structure at several viral genomic loci(especially regions encoding immediate-early and early genes), as well as a rapid change involving increasing H3ac and H3K4me3 marks and decreasing H3K27me3 upon reactivation (Toth et al., 2010). These results were later supported by the finding that RNA polymerase Ⅱ-mediated transcription of many lytic genes is paused at the elongation step during latency, but can be promptly reactivated by external stimuli (Toth et al., 2012). This bivalent control of gene expression is reminiscent of that observed in embryonic stem cells (Bernstein et al., 2006). Further experiments implicated EZH2, the H3K27me3 histone methyltransferase of the Polycomb group proteins(PcG), as a key regulator of this modification. The current view is that this epigenetic regulator contributes to the maintenance of latency (Toth et al., 2010), and promotes cancer angiogenesis (He et al., 2012).
The level of H3K9 methylation associated with viral episomes also appears to be tightly regulated. It has long been known that the terminal repeat(TR) region of KSHV episomes accumulates heterochromatin components, and that this is mediated by the latency-associated nuclear antigen(LANA) (Szekely et al., 1999; Mattsson et al., 2002; Lim et al., 2003; Viejo-Borbolla et al., 2003; Sakakibara et al., 2004). The genome-wide studies previously mentioned characterized the pattern of H3K9me3 across viral episomes during latency and lytic reactivation (Gunther and Grundhoff, 2010; Toth et al., 2010). The H3K9me3 demethylase, JMJD2A, was later shown to play a critical role in regulating this modification and, thus, the latent-lytic switch (Chang et al., 2011). The H3K9me1/2 histone demethylase, KDM3A/JMJD1A, has also been implicated in the control of latency, a function that likely depends on its association with LANA (Kim et al., 2013). Another mechanism by which LANA may contribute to epigenetic modifications of KSHV episomes is through interacting with Set1, the H3K4 methyltransferase (Hu et al., 2014). Intriguingly, K-bzip, an immediate-early gene product essential for viral lytic replication (Ellison et al., 2009; Lefort and Flam, 2009), was shown to inhibit JMJD2A demethylase activity (Chang et al., 2011). K-bzip has also been shown to modulate histone acetylation by interacting with and inhibiting histone deacetylases(HDACs) (Martinez and Tang, 2012). Several studies have documented the effect(s) of chemical HDAC inhibitors on gammaherpesvirus reactivation (Miller et al., 1997; Gwack et al., 2001; Lu et al., 2003; Ye et al., 2007; Gorres et al., 2014; Li et al., 2014; Lu et al., 2014; Shin et al., 2014; Yu et al., 2014), and some HDAC inhibitors have been explored as potential therapies to treat KSHV-related malignancies (Niedermeier et al., 2006; Bhatt et al., 2013).
Several recent studies investigated histone modifications following de novo infection of KSHV. The first, by Dr. Jae Jung's group, described a biphasic euchromatin-to-heterochromatin transition following infection (Toth et al., 2013b). Upon KSHV infection of endothelial cells, they found high levels of activating histone marks(H3K4me3 and H3K27ac) deposited on viral genomes at early times post-infection, which decreased at later times(24–72 h) concomitant with increasing repressive marks(H3K27me3 and H2AK119ub). Importantly, these epigenetic modifications were accompanied by corresponding changes in KSHV gene expression. In contrast, KSHV-infected epithelial cells adopt a transcriptionally active euchromatin state, resulting in expression of lytic genes (Toth et al., 2013b). Another study also profiled epigenetic modifications in endothelial cells following KSHV infection, and went on to characterize the role of nuclear domain 10(ND10) components during the establishment of latency (Gunther et al., 2014). A third group characterized the pattern of epigenetic modifications following KSHV infection of human peripheral blood mononuclear cells(PBMCs), which were correlated to the KSHV gene expression program (Jha et al., 2014). Taken together, these findings corroborate the views that (ⅰ) histone modifications of the KSHV genome correlate predictably with latent/lytic gene expression; and (ⅱ) cell type-specific epigenetic factors are a key determinant in deciding the fate of KSHV replication.
-
In the past decade, our understanding of the nuclear organization has broadened significantly, aided by advances in microscopy techniques and the development of chromosome confor-mation capture(3C) and its ChIP-based modifications (Gavrilov et al., 2009; Woodcock and Ghosh, 2010; de Witandde Laat, 2012). The National Institutes of Health have realized the importance of this field, and recently started the 4D Nucleome program, the aim of which is to underst and the spatial and temporal organization of the nucleus and how it relates to cellular processes during development or disease progression (NIH, 2014). A recent collaboration by our group and the lab of Dr. Jonathan Dennis assessed nucleosome redistribution at a subset of human genomic loci(genes involved in innate immunity) during a time course of lytic reactivation. Intriguingly, it was found that changes in nucleosome distribution in response to KSHV reactivation were widespread, transient, and directed by the underlying DNA sequence (Sexton et al., 2014a; Sexton et al., 2014b). A follow-up study using the same model system of KSHV reactivation analyzed nucleosome architecture at the transcription start sites of all human genes, and further demonstrated that changes in nucleosome distribution potentiated regulatory factor binding (Sexton et al., 2015).
KSHV episomes are tethered to the cellular chromatin by the major latent protein LANA through its interactions with histones and other cellular chromatin components. This raises an interesting question: is tethering to the host chromosome random, or could it be influenced by higher-order chromatin structure? Architectural proteins, including cellular chromatin boundary factor(CTCF) and cohesin, are crucial for sister chromatid cohesion and chromosome segregation (Merkenschlager and Odom, 2013). Work by Dr. Paul Lieberman's group and others has offered insights into the roles of these proteins during KSHV replication(reviewed in (Chen et al., 2013; Lieberman, 2013)). These studies suggest that CTCFcohesin facilitates stable episome maintenance (Stedman et al., 2008; Kang and Lieberman, 2011; Kang et al., 2011a), and is also involved in the regulation of transcription elongation and nucleosome position (Kang and Lieberman, 2011; Kang et al., 2013). 3C methods were used to show that CTCF-cohesin mediate the formation of a DNA loop between regulatory regions of latent and lytic genes (Kang et al., 2011a). It has been postulated that CTCF-cohesin association with CTCF-binding sites present at the boundaries of lytic gene promoters may serve the additional function of protecting their bivalent chromatin organization (Chen et al., 2012; Lieberman, 2013). Future investigation of higher-order chromatin structure with regard to KSHV will likely reveal new regulatory mechanisms affecting chromatin-dependent processes.
-
Non-coding RNAs(ncRNAs) have received increasing attention over the past decade, owing to their ubiquitous nature and fundamental roles in the regulation of DNA methylation, histone modifications, chromatin remodeling, and post-transcriptional regulation of gene expression. KSHV possesses a 1.1 kb long non-coding RNA(lncRNA) referred to as polyadenylated nuclear RNA(PAN RNA). Several groups contributed to the early characterization of PAN RNA, but only recently have its roles in KSHV gene expression and immune modulation been elucidated (Borah et al., 2011; Rossetto and Pari, 2011; Rossetto and Pari, 2012; Rossetto et al., 2013). These studies support the notion that, like cellular lncRNAs, PAN RNA may play a role in epigenetic regulation (Figure 2; reviewed in (Campbell et al., 2014; Rossetto and Pari, 2014)).
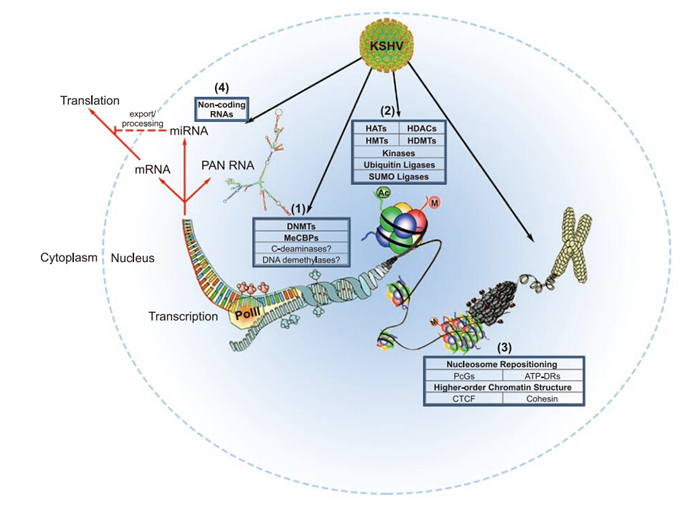
Figure 2. The KSHV Epigenome. Epigenetic regulation by KSHV affects both the viral and host genome, and can be classified into one of four major categories. (1) The state of DNA methylation affects nucleosomal structures. Regions with increased DNA methylation are often associated with transcriptional silencing. (2) Post-translational modifications (PTMs) of histone tails affect local chromatin structure and accessibility of transcription factors. Many different PTMs, including acetylation, methylation, phosphorylation, ubiquitination, and sumoylation, have been implicated in KSHV gene expression and the latent-lytic switch. (3) Nucleosome position and higher-order chromatin structure are intimately related to host chromatin-dependent processes, as well as KSHV episomal maintenance, transcriptional regulation, and control of latency. (4) Finally, non-coding RNAs, including viral/host miRNAs and KSHV PAN RNA, play critical roles in post-transcriptional epigenetic regulation. Importantly, these processes are inter-related, and all contribute to the regulation of transcription and subsequent RNA-dependent processes (denoted by red arrows). Acronyms: DNMTs – DNA methyltransferases; MeCBPs – methyl CpG binding proteins; HATs – histone acetyltransferases; HDACs – histone deacetylases; HMTs – histone methyltransferases; HDMTs – histone demethyltransferases; SUMO – small ubiquitin-like modifier; PcGs – Polycomb group proteins; ATP-DRs – ATP-dependent remodelers; CTCF – CCCTC-binding factor; PolII – RNA Polymerase Ⅱ. PAN RNA minimum free energy structure was generated using RNAfold software (Gruber et al., 2008). The KSHV capsid structure was adapted from (Xinghong Dai, 2015). This figure was adapted by permission from Macmillan Publishers Ltd on behalf of Cancer Research UK: British Journal of Cancer (Flanagan, 2007), © 2007
In addition, KSHV encodes at least 17 microRNAs(miRNAs) (Cai et al., 2005). Several recent studies have attempted to identify the viral and cellular targets of these miRNAs (Samols et al., 2007; Skalsky et al., 2007; Abend et al., 2010; Lu et al., 2010; Gottwein et al., 2011; Liang et al., 2011b; Lin and Ganem, 2011; Lin et al., 2011; Malterer et al., 2011; Bai et al., 2014; Feldman et al., 2014; Plaisance-Bonstaff et al., 2014; Yang et al., 2014; Forte et al., 2015; Quan et al., 2015), and in doing so have discovered their diverse roles in immune evasion, transformation, and control of latency. Many excellent reviews summarize the known roles of miRNAs in KSHV replication and pathogenesis (Ganem and Ziegelbauer, 2008; Lei et al., 2010; Liang et al., 2011a; Lin and Flemington, 2011; Ziegelbauer, 2011; Ramalingam et al., 2012; Toth et al., 2013a; Zhu et al., 2013; Haecker and Renne, 2014; Qin et al., 2014). Notably, the application of a technique that combines UV cross-linking with immunoprecipitation(CLIP) has allowed for the investigation of RNA-protein interactions (Ule et al., 2003; Ule et al., 2005; Jensen and Darnell, 2008; Darnell, 2010). Variations of this method have recently been used to determine the targetomes of viral and host miRNAs in KSHV-infected cells (Gottwein et al., 2011; Kang et al., 2011b; Haecker et al., 2012; Quan et al., 2015; Haecker and Renne, 2014). Continued improvement of CLIPbased methods and/or development of novel techniques should revolutionize our understanding of ncRNA-mediated epigenetic regulation.
-
Oncogenic viruses, such as KSHV, provide a unique system to study and manipulate cell plasticity (Iacovides et al., 2013). In vitro experiments have shown that KSHV is capable of transcriptionally reprogramming its host, consequently altering the infected cell's phenotype (Carroll et al., 2004; Hong et al., 2004; Wang et al., 2004; Cheng et al., 2011). Multiple studies have characterized KSHV gene expression in relation to epigenetic modifications, at various stages of the KSHV life cycle and in different cell lines(discussed above). A recent study described a unique program of KSHV gene expression in primary human lymphatic endothelial cells(LECs), which did not correspond to canonical latent or lytic replication (Chang and Ganem, 2013). These results further strengthen the supposition that there do exist cell type-dependent mechanisms which regulate KSHV gene expression.
Next-generation sequencing of RNAs(RNA-seq) can be used to identify and characterize mechanisms of transcriptional regulation. Recently, RNA-seq, in combination with sequencing the ribosome-protected RNAs(ribosome profiling or RIBO-seq), uncovered new genomic features and novel regulatory mechanisms at the level of transcription, RNA processing, and translation (Arias et al., 2014). The resulting annotation of the KSHV genome, termed KSHV 2.0, offers a comprehensive view of KSHV gene regulation during lytic reactivation. The application of RNA-seq to host transcriptional profiling of cells infected by KSHV is limited. Two studies have characterized the host transcriptome in cells exogenously expressing KSHV gene products: SOX (Clyde and Glaunsinger, 2011) or PAN RNA (Rossetto et al., 2013). There is currently only one published report, by Mercier et al., describing RNAseq analysis of host gene expression in KSHV-infected cells. They made the unexpected finding that LANA association does not lead to global host transcriptional remodeling (Mercier et al., 2014). While host transcriptome analysis of KSHV-infected cells is still in its infancy, it will likely have increasing applications in the study of KSHV in the coming years.
General overview: the significance of chromatin structure throughout the KSHV life cycle
Epigenetic regulation of KSHV gene expression
DNA methylation
Histone modifications and histone-modifying enzymes
Higher-order chromatin structure
Non-coding RNAs
Patterns of KSHV gene expression and host transcriptional reprogramming
-
The discovery of KSHV as the etiological agent of KS necessitated the development of model systems to study this virus in the context of its associated diseases. B cell lines from patients with PEL served as the first in vitro systems. These allowed for analysis of the viral genome and all aspects of the life cycle(latency, lytic reactivation, and infection). Furthermore, the ability to induce the production of viral proteins/particles facilitated the production of assays to detect KSHV seroprevalence in naturally infected populations. Endothelial cells also provided important in vitro models, because of their clear relevance to KS pathogenesis. In vivo, endothelial cells harbor KSHV genomes and are presumed to be the precursors of the spindle cells characteristic of KS lesions. Within the past decade, new and improved systems have served to enhance the efficiency of lytic reactivation and allow for manipulation of the viral genome, making in-depth functional studies possible. Animal models of KSHV-associated diseases have obvious benefits for studying KSHV pathobiology, but each also has its unique drawbacks. The following subsections will summarize the model systems that have been developed to study KSHV, and how they have contributed to our current knowledge of KSHV replication and pathogenesis. Many of the in vitro systems that have been used to study KSHV are compiled in Table 1.

Table 1. in Vitro Models to Study KSHV Replicatuib and Pathogenesis
-
Two cell lines derived from HIV/AIDS patients with body cavity-based lymphomas(AIDS-BCBLs) were established shortly following the discovery of KSHV (Cesarman et al., 1995). These cell lines, BC-1 and BC-2, were used to begin investigating the genetics of this newly described tumor virus (Cesarman et al., 1995). This was followed by the establishment of the BC-3 cell line, derived from primary effusion BCBLs, to address the problem of concomitant EBV infection in other cell lines (Arvanitakis et al., 1996). The first lytic reactivation system of KSHV in culture was achieved in 1996 by Renne et al., using BCBL-1 cells induced by TPA (Renne et al., 1996). Shortly thereafter, it was shown that HDAC inhibitors are also capable of inducing KSHV lytic reactivation (Miller et al., 1997). These early systems were vital to the characterization of KSHV latent/lytic replication, and provided viral antigens for serological assays (Gao et al., 1996; Kedes et al., 1996; Miller et al., 1996; Miller et al., 1997). A few years later, Cannon et al. established a new PEL cell line, JSC-1, which yielded two orders of magnitude more infectious virions compared to other PEL cells, but are also co-infected by EBV (Cannon et al., 2000).
-
Although PEL cell culture systems have served as vital systems for characterizing and propagating KSHV, it is also necessary to study KSHV infection in endothelial cells, as KS spindle cells are thought to be of endothelial origin (Weninger et al., 1999). KS cells express both lymphatic endothelial cell(LEC) and blood endothelial cell(BEC) markers (Wang et al., 2004). Both LECs and BECs are susceptible to KSHV infection, after which transcriptional reprogramming will result in BECs resembling LECs and vice versa (Carroll et al., 2004; Hong et al., 2004; Wang et al., 2004). When bone-marrow microvascular endothelial cells(BMECs) and human umbilical vascular endothelial cells(HUVECs) are infected with KSHV, both develop spindle cell-like morphology, as well as long-term proliferation and cell survival (Flore et al., 1998). Dermal microvascular endothelial cells(DMVECs) can support long-term latent KSHV infection with a small occurrence of cells spontaneously entering the lytic cycle, which is reminiscent of KS tumors (Panyutich et al., 1998; Moses et al., 1999). KSHVinfected DMVECs develop spindle shapes resembling KS and display transformed characteristics, including loss of contact inhibition and acquisition of anchorage-independent growth (Moses et al., 1999). However, a low percentage of cells are infected upon primary infection, and the infection does not spread throughout the culture, which may be due to the limited life span of DMVEC cultures.
DMVEC have been immortalized with hTERT to develop telomerase-immortalized microvascular endothelial(TIME) cells, a cell line that is susceptible to infection via cell-free KSHV (Lagunoff et al., 2002). TIME cells latently infected with KSHV can be reactivated with TPA, and resulting virions can successfully infect uninfected TIME cultures (Lagunoff et al., 2002). However, TIME cells cannot support long-term episomal maintenance (Lagunoff et al., 2002). Telomerase-immortalized human umbilical vein endothelial(TIVE) cells are also susceptible to KSHV infection and can be reactivated by RTA expression (An et al., 2006). TIVE cells were transformed by selecting cells that maintained KSHV episomes, resulting in TIVE long-term culture(LTC) (An et al., 2006). Unlike TIME cells, TIVE LTC cells support stable KSHV infection without selection and form tumors in nude mice (An et al., 2006). The tumor formations express the KS markers vascular endothelial growth factor(VEGF), basic fibroblast growth factor, and IL-6 (An et al., 2006).
In vivo, circulating KSHV has been found to primarily infect B cells (Ambroziak et al., 1995; Mesri et al., 1996). However, in vitro infection of B cells with soluble KSHV is often inefficient and abortive (Renne et al., 1998; Blackbourn et al., 2000; Bechtel et al., 2003). Myoung and Ganem were able to overcome this by co-culturing reactivated iSLK.219 cells with lymphoid cells. They successfully infected BJAB, Ramos, BCBL-1, JSC1, Jurkat, and SupT1 cells and found BJAB B cells and SupT1 T cells to be the most efficiently infected (Myoung and Ganem, 2011a). In a separate study, Myoung and Ganem also showed that KSHV is capable of infecting primary human tonsillar T cells, but could not maintain persistent infection or immortalize these cells (Myoung and Ganem, 2011c). Kati et al. developed a novel method of reactivating KSHV in latently infected B cells by exposing IgM-expressing BrK.219 cells, a BJAB cell line latently infected with rKSHV.219, to anti-IgM (Kati et al., 2013). Another more recent study analyzed KSHV/EBV-negative B cell lines for their permissiveness to KSHV infection (Dollery et al., 2014). They identified MC116, a B cell line expressing IgMλ, as susceptible to KSHV infection, and were able to induce virion production with butryate or anti-IgM (Dollery et al., 2014). This result is not surprising, as a previous study found that KSHV almost exclusively infects a subset of B cells expressing IgMλ, which comprises about 20% of human tonsils (Hassman et al., 2011).
-
The discovery of KSHV RTA as the master lytic switch protein(its ectopic expression is sufficient to induce lytic reactivation Sun et al., 1998) presented the possibility to engineer systems capable of robust and efficient induction of the lytic cycle. Nakamura et al. developed a Flp-In-mediated recombination and tetracycline-inducible expression system in KSHV-infected PEL cells(TREx BCBL1-Rta), which can be utilized to study individual KSHV genes and was more efficient than previous methods of induction (Nakamura et al., 2003). In the time since, several studies have utilized this system to study KSHV lytic replication.
Despite the development of numerous useful cell culture systems, a major caveat that remained was the high rates of spontaneous lytic replication, which made it difficult to accurately study latent viral gene expression (Myoung and Ganem, 2011b). To address this issue, Myoung et al. developed iSLK.219 cells, in which KSHV latency is tightly controlled, but lytic reactivation is efficiently inducible by doxycycline (Myoung and Ganem, 2011b). It is important to note that iSLK and iSLK.219 cell lines were developed from SLK cells, which were initially described as being of endothelial origin (Herndier et al., 1994), but were recently reported to have been contaminated with Caki-1 cells of renal carcinoma origin (Sturzl et al., 2013). This finding calls into question the use of SLK cells as a model for KS.
-
A major leap forward in the development of KSHV cell culture models was the use of bacterial artificial chromosomes(BACs) containing the KSHV genome isolated from BCBL-1 cells (Zhou et al., 2002). The construction of BAC36 was integral to the study of KSHV, as it provided an efficient method of primary infection in cultured cells (Zhou et al., 2002). This system also allowed for more efficient primary infection and precise studies of viral genes, since BAC recombineering could be performed to mutate theoretically any gene of interest. Within the decade after its description, several studies utilized BAC36 to study KSHV infection and replication, as well as the functions of specific viral products (Gao et al., 2003; Luna et al., 2004; Ye et al., 2004; Xu et al., 2006; Zhu et al., 2006; Brinkmann et al., 2007; Majerciak et al., 2007; Yoo et al., 2008; Li and Zhu, 2009; Lu et al., 2010; Sathish and Yuan, 2010; Budt et al., 2011; Lin et al., 2011; Yakushko et al., 2011; Cho and Kang, 2012; Hyosun Cho, 2012; Lu et al., 2012; Martinez and Tang, 2012; Rossetto and Pari, 2012; Peng et al., 2014; Walker et al., 2014). Importantly, it was reported that a region of the KSHV genome(including part of ORF19 and all of ORFs 18, 17, 16, K7, K6, and K5) was duplicated in BAC36 (Yakushko et al., 2011). Recently, a new KSHV BAC was developed, named BAC16 (Brulois et al., 2012). BAC16 was derived from rKSHV.219, which was isolated from KSHV and EBV co-infected JSC-1 cells(Vieira and O′Hearn, 2004). Therefore, BAC16 expresses red fluorescent protein(RFP) as a marker of lytic infection, in addition to GFP, which is expressed in both BAC36 and BAC16 latently-infected cells (Zhou et al., 2002; Brulois et al., 2012). Furthermore, induction of lytic reactivation in BAC16 stably transfected iSLK cells(iSLK.BAC16) yields higher titers of infectious virions than previous systems.
-
As the evidence of KSHV's causative role in KS grew, it became necessary to develop an animal model to allow for systemic studies. One of the major obstacles in developing an animal model has been the low incidence of KS/PEL-like disease symptoms despite high susceptibility to KSHV infection. One of the first animal models utilized was severe combined immunodeficient(SCID) mice that had been injected with KSHV-infected BCBL-1 cells, similar to what had been done with EBV and SCID mice (Rochford and Mosier, 1995; Picchio et al., 1997). Tumors were able to form upon injection of KSHV infected BCBL-1 cells, however researchers were unable transmit virus to PBMC grafts (Picchio et al., 1997). This was followed by the observation of persistent infection with PEL-derived KSHV and viral replication in SCID-hu THY/LIV mice (Dittmer et al., 1999). CD19+ lymphocytes were permissive to infection, although there was no morphological change of the implant(no cytopathic effects) (Dittmer et al., 1999). Since then, KSHV models have been attempted with Rhesus macaques, marmosets, and mice. The MHV-68 model can result in lymphoproliferative disease, but not KSlike symptoms(Sunil-Chandra et al., 1994). In Rhesus macaques, co-infection with RRV and SIV results in the development of lymphoma at a similar rate(20–30%) as that seen in AIDS-KS patients (Orzechowska et al., 2008). The marmoset model developed by Chang et al. develops an antibody response that lasted up to 1.5 years following intravenous infection with rKSHV.219 (Chang et al., 2009). However, viral recovery from the PBMCs of the infected marmosets was not successful, and the anti-KSHV response to oral infection was not as robust (Chang et al., 2009).
Multiple mouse models have been developed and studied within the last year. Wang et al. have attempted to infect the humanized-bone marrow, liver, thymus(huBLT) model with rKSHV.219 via natural routes, such as oral mucosa, since transmission of KSHV has been found to occur primarily via saliva and to reside in the tonsils (Vieira et al., 1997; Mayama et al., 1998; Chagas et al., 2006). This model may represent a useful system for investigating transmission routes (Wang et al., 2014). They found that the mice were successfully infected, resulting in latent/lytic replication in B cells and macrophages of the spleen and latent infection of macrophages in the skin (Wang et al., 2014). However, an immune response was absent when tested three months post-infection (Wang et al., 2014). Ashlock et al. developed a mouse model by transfecting murine bone marrow-derived endothelial lineage cells with BAC36(mECK36 cells) and injecting them into immunodeficient mice, which induced the formation of KS-like tumors. However, these mice were not able to produce progeny virions (Ashlock et al., 2014). When they replaced BAC36 with rKSHV.219, KS-like tumors were accompanied by the production of herpesvirus-like particles, as observed by electron microscopy (Ashlock et al., 2014). Additionally, treatment of mice with an HDAC inhibitor induced lytic reactivation in vivo, indicating this could be a useful system to test antiviral treatments (Ashlock et al., 2014). A xenograft mouse model of KSHV-associated PEL has been previously used to study the effects of host environment on tumor growth, and to test the efficacy of PEL therapies (Staudt et al., 2004; Sin et al., 2007; Bhatt et al., 2010; Sarosiek et al., 2010; Towata et al., 2010; Bhatt et al., 2013). However, the degree to which these tumor formations were related to natural occurring PEL had not previously been investigated. In a recent study, Dai et al. injected BCBL-1 cells into non-obsess diabetic/severe combined immunodeficient mice(NOD/SCID) (Dai et al., 2014). This resulted in rapid tumor growth, massive ascites, and splenic enlargement within 3–4 weeks post-infection. However, a multitude of tumor and symptomatic characteristics differed from what is observed in PEL patients (Dai et al., 2014). Recent reports that KSHV can efficiently infect and transform primary rat embryonic metanephric mesenchymal precursor(MM) cells suggests that this system could be useful in study mechanisms of KSHV-induced growth deregulation and oncogenesis (Jones et al., 2012; Moody et al., 2013; Liang et al., 2014). However, it was recently shown that the GFP marker expressed from recombinant herpesviral genomes may not be a reliable marker for infection in this system (Ellison and Kedes, 2014). This highlights the notion that when evaluating new model systems for the study of KSHV pathogenesis, thorough analyses must be performed to avoid misinterpretation of data.
Patient-derived cell lines as model systems in initial studies of KSHV
Endothelial and B cell models
Inducible control of RTA
Genetic manipulation of KSHV genome
In Vivo model systems
-
Our current understanding of the KSHV epigenome can be attributed, in large part, to innovations in molecular biology tools and sequencing technology. Moreover, the development and applications of cell culture and animal model systems have made it possible to study every facet of KSHV pathobiology. These systems have facilitated the detailed investigation of the two alternative life cycles of KSHV and how they contribute to pathogenesis, and unveiled the critical roles of epigenetic phenomena. With each result, we add pieces to a puzzle that is becoming simultaneously clearer and exceedingly complex. In our attempts to better underst and KSHV, we have shed light on the molecular mechanisms that govern fundamental cellular processes. Future studies aiming to elucidate the relationships between epigenomic marks(DNA methylation, histone modifications, nucleosome position, and higher-order chromatin structure) and chromatin-dependent processes(replication, recombination, repair, and transcription) should yield exciting insights into the biology of KSHV and its human host.
-
We would like to apologize to the authors of many important studies that have not been cited due to space limitations. This work was supported by National Institutes of Health grant R01DE016680 to Fanxiu Zhu and F31CA183250 to Denis Avey. We thank Sarah Tepper for critical reading of the manuscript and helpful edits.
-
The authors declare that they have no conflict of interest. This article does not contain any studies with human or animal subjects performed by any of the authors.

 DownLoad:
DownLoad: