










HTML
-
Human immunodeficiency virus type Ⅰ(HIV-1) viral protein R(Vpr)is a 96-amino-acid, 14-kDa accessory protein that plays an important role in viral replication(Romani and Engelbrecht, 2009). Vpr is expressed in the late phase of the HIV-1 lifecycle and packaged into virions through interaction with the p6 domain of the Gag protein(Cohen et al., 1990; Muller et al., 2000). Many functions have been reported for Vpr, including promoting reverse transcription, participating in nuclear transport of the pre-integration complex(PIC), enhancing viral and cellular gene transcription through activating the NF-κB pathway, disruption of cell cycle control, and the induction of apoptosis(Yedavalli et al., 2005; Romani and Engelbrecht, 2009).
The most extensively studied function of Vpr is the induction of cell cycle arrest at the G2 to mitosis transition(G2/M)in most cycling eukaryotic cells. The G2/M transition in mammalian cells is controlled by the cell division cycle 2(Cdc2):CyclinB1 complex(Romani and Engelbrecht, 2009). This complex is inactivated when Cdc2 is phosphorylated on Tyr14 and Tyr15, and is activated when Cdc25C dephosphorylates these two threonine residues(Ohi and Gould, 1999). It has been reported that Vpr binds to Cdc25C and inactivates the phosphatase activity of Cdc25C(Goh et al., 2004; Kino et al., 2005). Cyclin-dependent kinase inhibitor p21/Waf1/Cip1 also inhibits G2/M phase transition, and can be induced by Vpr at the transcription level(Chowdhury et al., 2003; Vazquez et al., 2005). It has been shown that interaction between Vpr and the DDB1-Cullin4A-VPRBP E3 ubiquitin ligase complex is required for the induction of G2/M arrest(Belzile et al., 2007; Hrecka et al., 2007). Recently, using HeLa cells, Laguette et al. reported that Vpr usurps the structure-specific endonuclease(SSE)regulator SLX4 complex(SLX4com)to promote G2/M cell cycle arrest(Laguette et al., 2014). These different mechanisms behind the ability of Vpr to induce G2/M arrest reflect the involvement of Vpr in multiple cellular pathways(Romani and Engelbrecht, 2009).
Previous studies have shown that Vpr activates the NF-κB pathway as well as enhancing transcription from the HIV-1 long terminal repeats(LTR)promoter(Subbramanian et al., 1998; Roux et al., 2000; Varin et al., 2005; Hoshino et al., 2010). Our group recently reported that Vpr interacts with IKKα/β and induces phosphorylation of p65 and p100, which leads to activation of the NF-κB pathway. Knockdown of IKKα/β, p65, or p100 in HeLa cells impairs the induction of the NF-κB reporter as well as the HIV LTR reporter gene by Vpr(Liu et al., 2013). Given the role of NF-κB in many cellular processes, including cell cycling(Ledoux and Perkins, 2014), there might be a connection between the two functions of Vpr in activating NF-κB and inducing G2/M cell cycle arrest.
To test this hypothesis, we studied a panel of Vpr mutants for their ability to activate the NF-κB pathway and induce G2/M arrest. In order to facilitate comparison with our previous data and the results from other laboratories, this study was carried out using HeLa cells. The results showed that Vpr mutants deficient in NF-κB activation also failed to induce G2/M cell cycle arrest, which suggests that activation of the NF-κB pathway by Vpr might contribute to Vpr-induced G2/M cell cycle arrest. In order to test this, the key factors in the NF-κB pathway were further depleted and partially rescued Vpr-induced G2/M cell cycle arrest. Together, our data indicate that Vpr induces G2/M cell cycle arrest at least partially by activating the NF-κB pathway.
-
The DNA constructs 3×NF-κB-Luc, Flag-Vpr, and pQC-Flag-Vpr have been described previously(Liu et al., 2013; Liu et al., 2014). Flag-Vpr mutant constructs were generated using the site-directed polymerase chain reaction(PCR)mutagenesis method, and all mutations were verified by DNA sequencing.
-
HeLa and HEK293T cells were maintained in Dulbecco's modified Eagle's medium(DMEM)supplemented with 10% fetal bovine serum and 100 U/mL penicillin/streptomycin. Cells were maintained in a humidified atmosphere containing 5% CO2 at 37 ℃. Cells were transfected by PEI(Polysciences, Warrington, USA)or Lipofectamine 2000(Invitrogen, Carlsbad, USA)according to the manufacturers' instructions.
-
The generation of p65, RelB, IKKα, IKKβ, and IKKαβ knockdown HeLa cell lines has been previously described(Liu et al., 2013). Briefly, shRNA oligos were designed to target the above genes and cloned into a pSIREN-RetroQ vector(Clontech, Mountain View, USA).pSIREN-RetroQ shRNA retroviral particles and pQC-Flag-Vpr retroviral particles were generated by transfecting HEK293T cells with pSIREN-RetroQ shRNA constructs or pQC-Flag-Vpr, together with pMLV-Gag-Pol and pVSV-G plasmids. At 48 h after transfection the viral supernatants were collected and centrifuged at 1, 000 × g for 15 min to remove residual cells and debris. The HeLa cells were then transduced by spinoculation with retroviral particles at 500 × g for 30 min at room temperature in the presence of polybrene(5 μg/mL). Stably transduced cell lines were selected with puromycin(2 μg/mL).
-
HeLa cells were transfected with the Flag-Vpr plasmid DNA. After 48 h the cells were rinsed twice with cold phosphate-buffered saline(PBS), then fixed with 70% ethanol at 4 ℃ overnight. After centrifugation at 500 × g to remove supernatants, the cells were rinsed with cold PBS and incubated in PBS containing 100 μg/mL RNase A for 30 min at 37 ℃. Cells were then stained with 50 μg/mL propidium iodide(PI)at room temperature for 15 min, and then analyzed by flow cytometry using BD FACSCalibur(BD Biosciences, San Diego, USA). Then the ModFit LT was used to analyze the cell cycle profiles. The(G2+M)/G1 ratios were calculated by dividing the proportion of cells in G2/M by the proportion of cells in G1.
-
HeLa cells were seeded into 12-well plates at a concentration of 1 × 105 cells/well. The following day, cells were transfected with 200 ng 3×NF-κB-Luc plasmid and 200 ng Flag-Vpr mutant plasmids along with 10 ng Renilla luciferase plasmid, then after 48 h the cells were harvested in lysis buffer and subjected to luciferase assay using the Dual-Luciferase reporter assay system(Promega, Madison, USA). The relative luciferase activity was calculated by dividing firefly luciferase activity by Renilla luciferase activity.
Plasmids
Cell culture and transfection
RNA interference
Cell cycle analysis
Luciferase assay
-
Unlike the other functions of Vpr, the amino acid determinants for activating the NF-κB pathway have not been well characterized. We therefore generated a panel of Vpr mutants that have been previously reported to impair Vpr functions(Figure 1A). The first group includes A30L, A59P, Q65A, H71C, R73A, G75A, S79A, and R80A, which impair the G2/M arrest activity of Vpr(Di Marzio et al., 1995; Mahalingam et al., 1997). The second group, A59P, H71C, R73A, R77A, R77Q, and R80A, fail to induce apoptosis(Jacotot et al., 2000; Patel et al., 2002). A30L impairs Vpr packaging into viral particles(Di Marzio et al., 1995). L64P and L67S mutants are defective in Vpr nuclear export(Sherman et al., 2001; Sherman et al., 2003). Residues S28, S79, S94, and S96 undergo phosphorylation(Zhou and Ratner, 2000; Agostini et al., 2002). The plasmid DNA of these Vpr mutants was first transfected into HeLa cells and their expression levels were examined by western blotting. The results showed that these mutants were expressed at similar levels(Figure 1B).

Figure 1. Activation of NF-κB by different Vpr mutants. (A) The diagrammatic structure of Vpr protein. Both the linear (upper) and the three-dimensional (lower) structure are shown. Vpr contains a flexible N-terminal domain (NTD), three α-helical domains, and a flexible C-terminal domain (CTD). The amino acid residues selected for mutation are indicated in red. The protein expression of WT and mutant Vpr was monitored by western blotting and is shown in (B). HeLa cells were transfected with p3×NF-κB-luc luciferase reporter plasmid and Flag-Vpr (WT or mutants) plasmid. Luciferase activities were measured 48 h after transfection. Relative luciferase activity (Rel. Luc. Act.) is shown in (C). Error bars represented SD of the means from three independent experiments.
First, the abilities of these Vpr mutants to activate the NF-κB pathway were measured. To this end a reporter construct called p3×NF-κB-luc was used, which has the firefly luciferase expressed from a promoter that has three binding sites for NF-κB. As an internal control to normalize transfection efficiency, the TK-RL plasmid that expresses Renilla luciferase from the thymidine kinase promoter was also co-transfected. The results in Figure 1C show that wildtype Vpr increased firefly luciferase expression fourfold, indicating a fourfold activation of the NF-κB pathway. Among the Vpr mutants tested, A30L, A30F, A59P, H71C, R73A, S79A, and R80A were unable to activate the NF-κB pathway.
Next the effects of these Vpr mutants on G2/M cell cycle arrest were measured. Cells were stained with propidium iodide(PI) and cell cycle profiles were calculated using the flow cytometry data. The expression of wildtype Vpr increased the percentage of G2 phase cells from 12.66% to 51.53%(Figure 2A). Different Vpr mutants exhibited various effects on G2/M arrest, ranging from wildtype Vpr-like activity(S28A, L67S, G75A, R77A, R77Q, and S94G/S96P)to moderately impaired activity(L64P and Q65A), severely impaired activity(A30L and A59P), and loss of activity(A30F, H71C, R73A, S79A, and R80A). This is the first comprehensive investigation of the amino acid residues in Vpr that are involved in activating the NF-κB pathway. Comparing the results of Figure 2A to those of Figure 1C reveals that the mutations A30L, A30F, A59P, H71C, R73A, S79A, and R80A impaired the ability of Vpr to activate the NF-κB pathway as well as to induce G2/M cell cycle arrest. This coincidence suggests that either both functions of Vpr involve the same protein structure or one function is a prerequisite for the other.
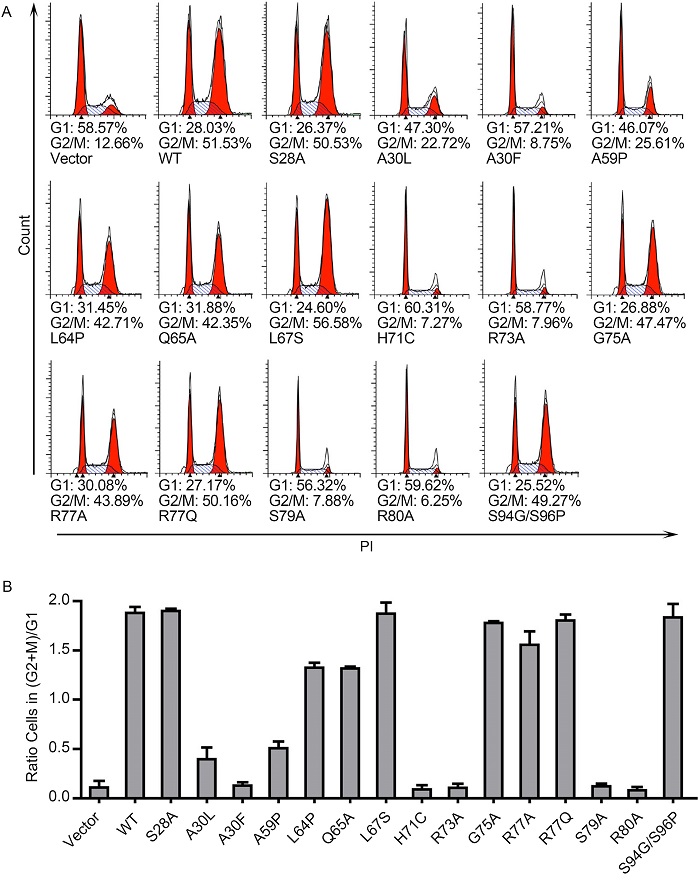
Figure 2. Induction of G2/M arrest by different Vpr mutants. HeLa cells were transfected with indicated Flag-Vpr mutants and harvested 48 h later. Cells were stained with PI and subjected to flow cytometry to measure DNA content. The represented cell cycle profiles are shown in (A), and the (G2+M)/G1 ratios in (B).
-
Our hypothesis was that if activation of the NF-κB pathway is the prerequisite for inducing G2/M arrest, then blocking the NF-κB pathway should attenuate the ability of Vpr to induce G2/M arrest. To test this hypothesis, p65 or RelB was first depleted by RNAi to block the NF-κB pathway and then Vpr was expressed. Cell cycle profiles were determined as described above. The results showed that knockdown p65 or RelB reduced the Vpr-induced G2/M cell cycle arrest by respectively 45% and 60%(Figures 3A–3C).
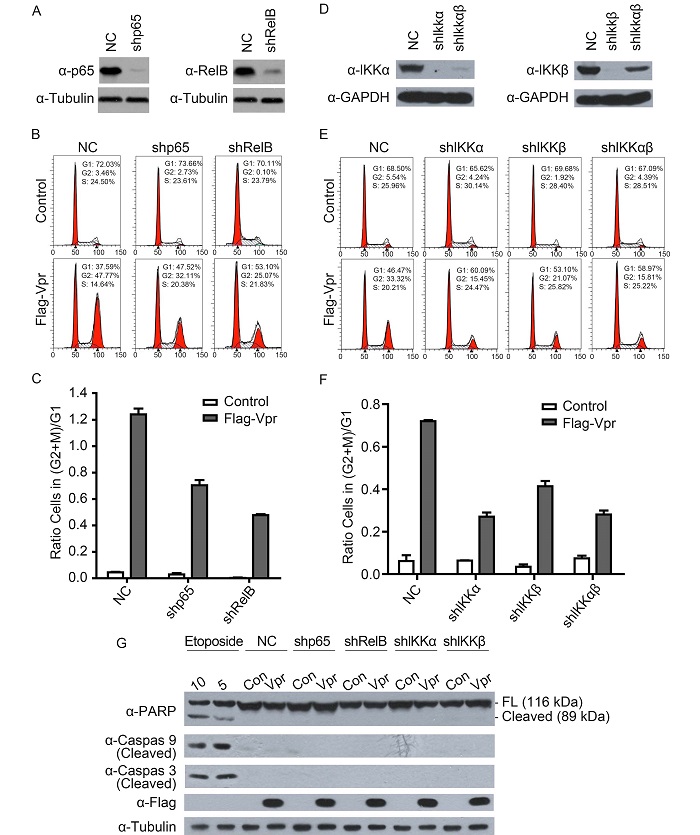
Figure 3. Vpr-induced cell cycle arrest in the NF-κB-downregulated HeLa cell lines. Western blotting analysis of p65 and RelB protein expression in shp65 and shRelB cell lines is shown in (A), and IKKα and IKKβ expression in shIKKα, shIKKβ and shIKKαβ cell lines in (D). HeLa knockdown cell lines were transduced with VSV-G pseudotyped retroviral particles expressing Flag-Vpr and harvested 48 h later. The cells were stained with PI and subjected to flow cytometry to measure DNA content. Cell cycle profiles are shown in (B) and (E). The (G2+M)/G1 ratios are shown in (C) and (F). (G) HeLa knockdown cell lines were transduced with retroviral particles that express Flag-Vpr or the vector control. After 48 h the cells were lyzed and subjected to western blotting with the indicated antibodies. As a positive control for apoptosis, cells were treated with 10 μmol/L or 5 μmol/L etoposide for 24 h. NC, negative control; FL, full length.
Our previous study showed that Vpr modulated the phosphorylation of p65 and p100 via its association with upstream kinases IKKα/β, which leads to the activation of both canonical and noncanonical NF-κB pathways(Liu et al., 2013). IKKα and IKKβ in HeLa cells were therefore further depleted and the effect on Vpr-induced G2/M arrest measured. Again, a significant attenuation in the ability to induce G2/M arrest was observed when either IKKα or IKKβ was knocked down(Figures 3D–3F).
The NF-κB pathway is also involved in apoptosis and G1/S phase transition, which can be monitored by analyzing cell cycle profiles. As shown in Figures 3B and 3E, the profiles of the G1 and S phases in the NF-κB-downregulated cell lines were similar to those of the negative control cells. Cells with a DNA content < 2N(sub-G1 cells)are usually the result of apoptotic DNA fragmentation. No sub-G1 peaks in the cell cycle profiles were observed(Figures 3B, 3E), which suggests that no apoptosis had occurred. Levels of the cleaved markers of apoptosis PARP, Caspase 3, and Caspase 9 were also measured by western blotting. As shown in Figure 3G, cells that were treated with the DNA damage agent etoposide underwent apoptosis; in contrast, the NF-κB-downregulated cells were not apoptotic with or without Vpr expression. These data indicate that downregulation of NF-κB does not cause G1/S arrest or apoptosis.
Taken together, these data demonstrate that activating the NF-κB pathway with Vpr contributes to the induction of the G2/M cell cycle arrest.
NF-κB activation and G2/M arrest induced by Vpr mutants
Activation of the NF-κB pathway promotes Vpr-induced cell cycle arrest
-
HIV-1 benefits from the arrest of the G2/M cell cycle by Vpr as the transcription levels of viral genes are the highest at G2 phase(Goh et al., 1998; Gummuluru and Emerman, 1999). This high level of viral gene transcription is partially attributable to the activation of the NF-κB pathway by Vpr. Our study further shows that activation of the NF-κB signaling pathway also assists Vpr in inducing G2/M arrest. This conclusion is based on two major pieces of evidence. First, Vpr mutants deficient in NF-κB activation also failed to induce G2/M cell cycle arrest. Second, blocking the NF-κB pathway by deleting the key factors p65, RelB, and IKKα/β dampened the ability of Vpr to induce G2/M arrest.
Several mechanisms have been reported by a number of studies that might highlight how Vpr causes G2/M arrest. Recently, Laguette et al. reported that Vpr prematurely activates SLX4com through recruiting the DDB1-Cullin4A-VPRBP E3 ubiquitin ligase complex and kinase-active PLK1. The activated SLX4-associated MUS81-EME1 endonucleases cleave DNA, causing replication stress, which halts cell cycle transition from G2 to M(Laguette et al., 2014). Replication stress often triggers ATR-CHK1 signaling, inhibition of the Cdc25C and Cdc2:CyclinB1 complex, and ultimately G2/M arrest(Bregnard et al., 2014). In addition to this mechanism, it has also been reported that Vpr can bind to Cdc25C(Goh et al., 2004; Kino et al., 2005)or induce the expression of cyclin-dependent kinase p21/Waf1/Cip1(Chowdhury et al., 2003; Vazquez et al., 2005). Both activities can lead to G2/M arrest.
Activation of the NF-κB pathway may promote G2/M arrest by directly or indirectly modulating the activity of the Cdc2:CyclinB1 complex. For example, it was reported that the expression of p21 can be stimulated by NF-κB(Seitz et al., 2000; Bren et al., 2001; Wuerzberger-Davis et al., 2005). It is also possible that NF-κB may overlap with DDB1-Cullin4A-VPRBP, SLX4com, and even the Cdc2:CyclinB1 pathways. In addition, IKK and NF-κB regulate the expression of CyclinD1 that controls G1 to S progression(Cao et al., 2001). IKKα does so by associating with and phosphorylated CyclinD1 and controlling the subcellular localization and proteolysis of CyclinD1(Kwak et al., 2005). Zhu et al. reported that IKKα prevents hypermethylation of 14-3-3σ, a G2/M checkpoint gene(Zhu et al., 2007). Through binding to IKK and activating the NF-κB pathway, Vpr may indirectly modulate the functions of the above regulators of cell cycle transition and thereby cause G2/M arrest.
Another function attributed to Vpr is the induction of apoptosis. Two recent studies show that in astrocytes Vpr induces the expression of IL-6, IL-8(Gangwani and Kumar, 2015), and CCL5(Gangwani et al., 2013), which are also mediated via the NF-κB pathway. These proinflammatory cytokines may play some roles in Vpr-induced apoptosis, which suggests that activation of the NF-κB pathway may also contribute to the other functions of Vpr.
In summary, we here report the interrelationship betw-een two important activities of Vpr, i.e. inducing G2/M arrest and activating the NF-κB pathway. Our data support the scenario that the activation of NF-κB by Vpr promotes G2/M arrest. This mechanism at least complements the others, including activating SLX4com though recruiting DDB1-Cullin4A-VPRBP(Laguette et al., 2014). Further study is warranted to investigate the involvement of the NF-κB pathway in cell cycle progression.
-
This work was supported by grants from the Chinese Ministry of Health(2012ZX10001006), the National Natural Science Foundation of China(81271812 and 31370182), 111 Project(B08011), and the Postgraduate Scholarship Program of the China Scholarship Council.
-
All the authors declare that they have no competing interests. This article does not contain any studies with human or animal subjects performed by any of the authors.
-
WQ conceived the study. ZL analyzed the data and wrote the manuscript. ZL, RL and YL performed the experiments. CL, JT, and WQ supervised the experimental work and participated in preparing the manuscript.

 DownLoad:
DownLoad: